

Cis-perturbation of cancer drivers by the HTLV-1/BLV proviruses is an early determinant of leukemogenesis
- PMID: 28534499
- PMCID: PMC5457497
- DOI: 10.1038/ncomms15264
Cis-perturbation of cancer drivers by the HTLV-1/BLV proviruses is an early determinant of leukemogenesis
Abstract
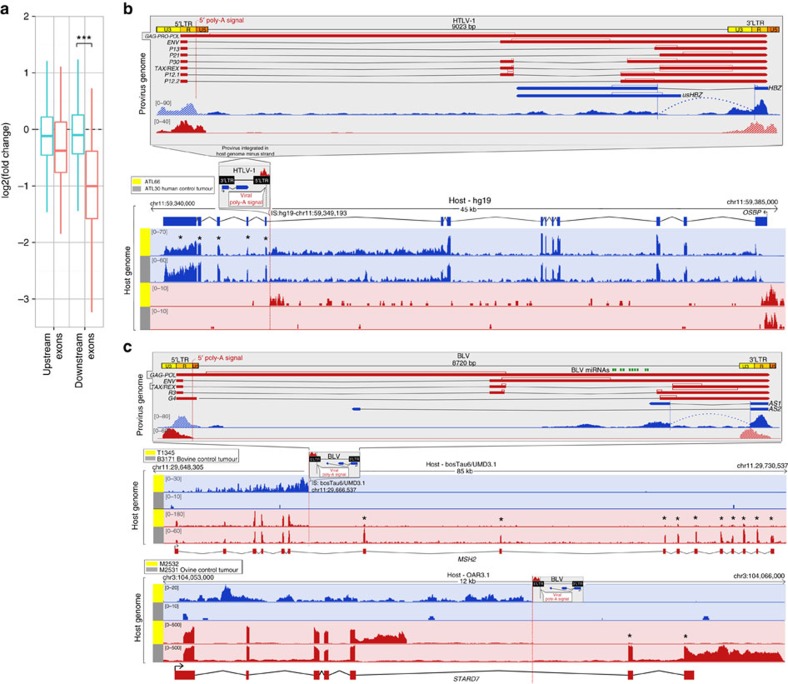
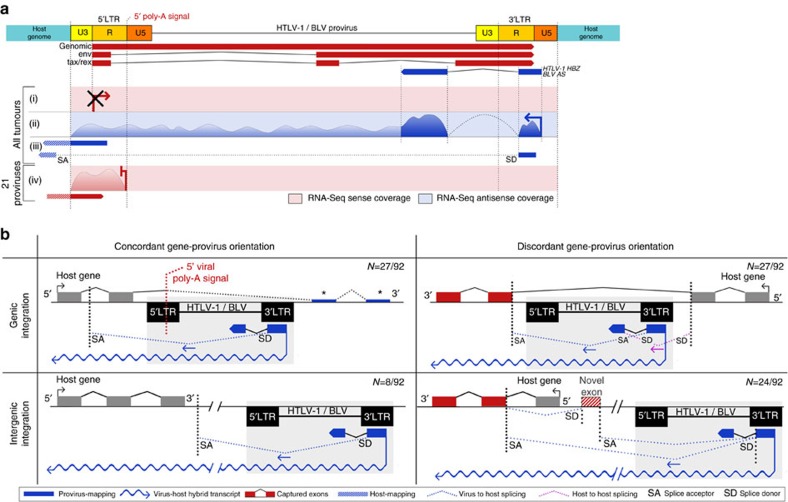
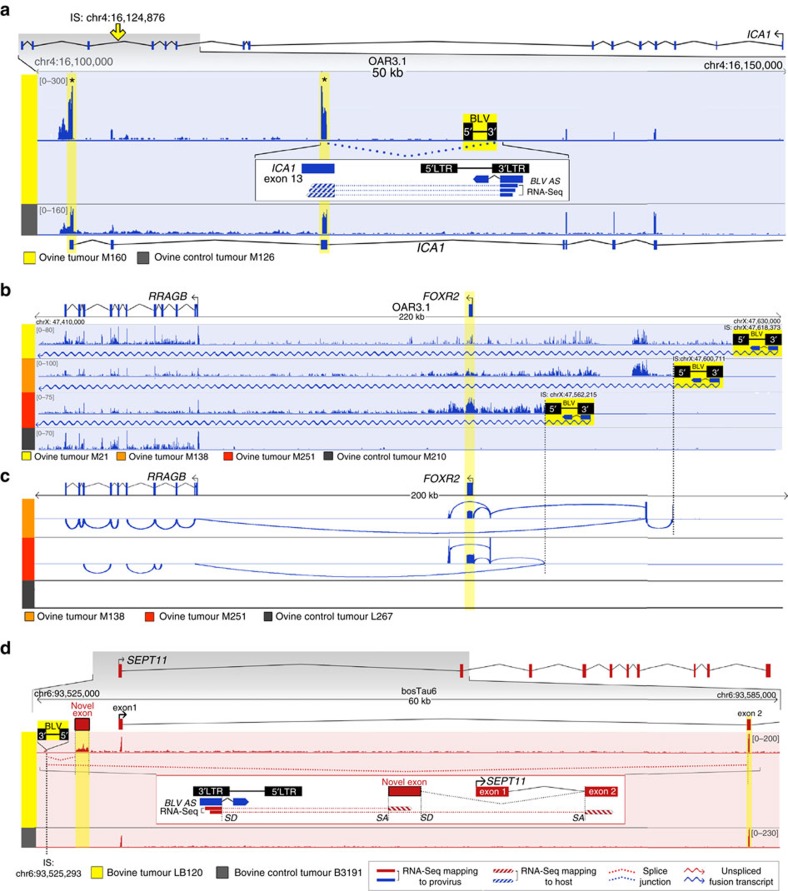
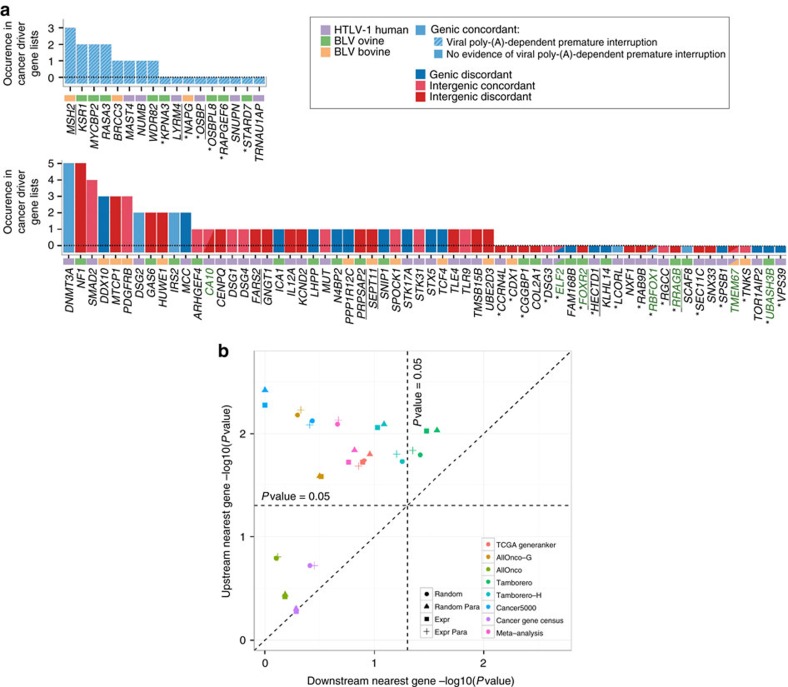
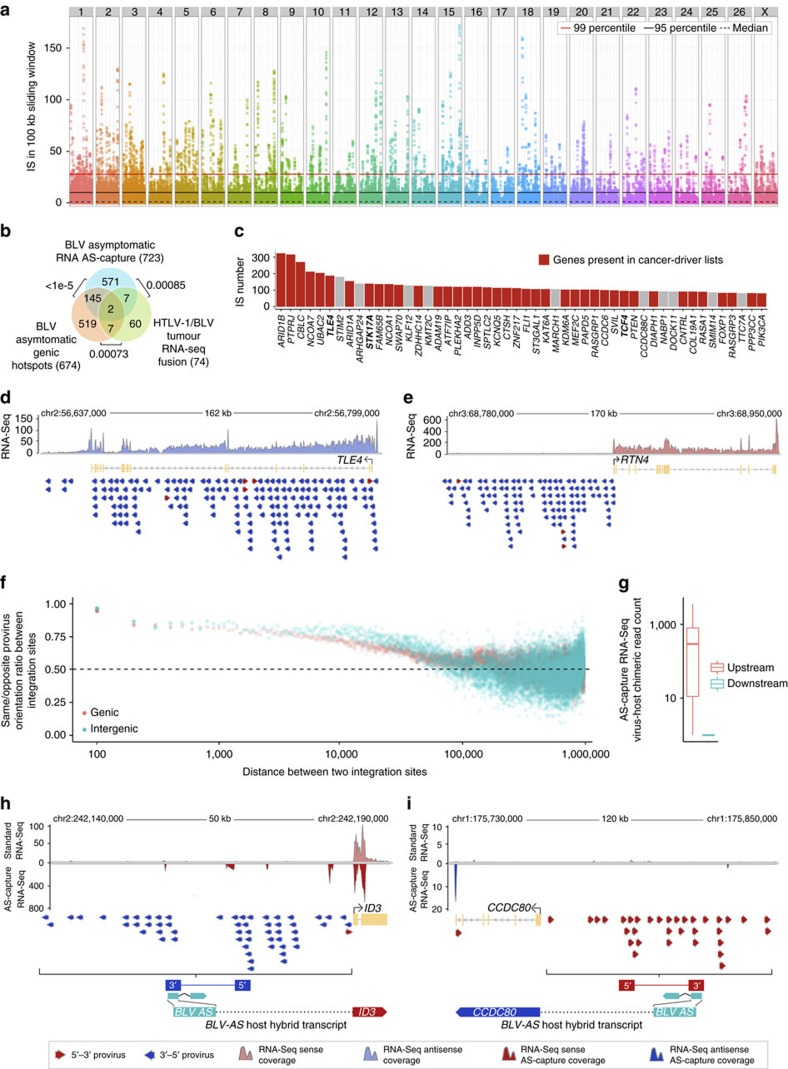
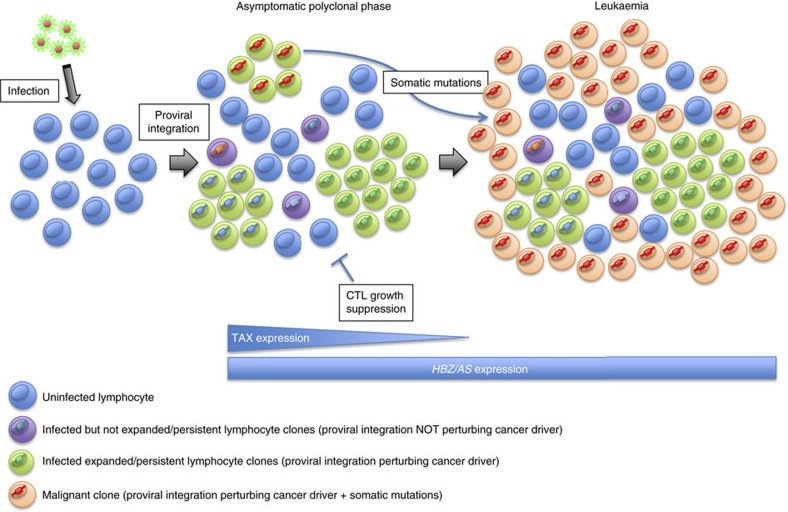
Human T-cell leukaemia virus type-1 (HTLV-1) and bovine leukaemia virus (BLV) infect T- and B-lymphocytes, respectively, provoking a polyclonal expansion that will evolve into an aggressive monoclonal leukaemia in ∼5% of individuals following a protracted latency period. It is generally assumed that early oncogenic changes are largely dependent on virus-encoded products, especially TAX and HBZ, while progression to acute leukaemia/lymphoma involves somatic mutations, yet that both are independent of proviral integration site that has been found to be very variable between tumours. Here, we show that HTLV-1/BLV proviruses are integrated near cancer drivers which they affect either by provirus-dependent transcription termination or as a result of viral antisense RNA-dependent cis-perturbation. The same pattern is observed at polyclonal non-malignant stages, indicating that provirus-dependent host gene perturbation contributes to the initial selection of the multiple clones characterizing the asymptomatic stage, requiring additional alterations in the clone that will evolve into full-blown leukaemia/lymphoma.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Ishitsuka K. & Tamura K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 15, e517–e526 (2014). - PubMed
-
- Burny A. et al. in Viruses and Cancer (eds Minson, A. C., Neil, J. C. & McRae, M. A.) 213–234 (Cambridge University Press, 1994).
-
- Bartlett P. C. et al. Options for the control of bovine leukemia virus in dairy cattle. J. Am. Vet. Med. Assoc. 244, 914–922 (2014). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
