

Gene expression variability and the analysis of large-scale RNA-seq studies with the MDSeq
- PMID: 28535263
- PMCID: PMC5737414
- DOI: 10.1093/nar/gkx456
Gene expression variability and the analysis of large-scale RNA-seq studies with the MDSeq
Abstract
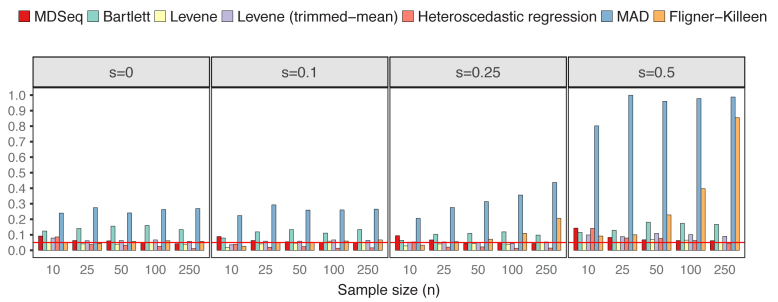
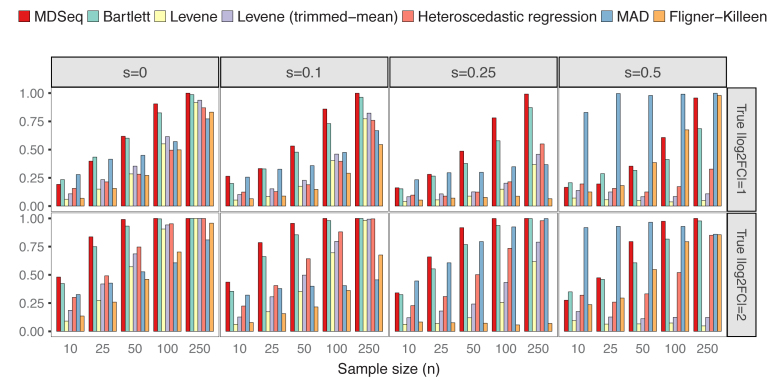
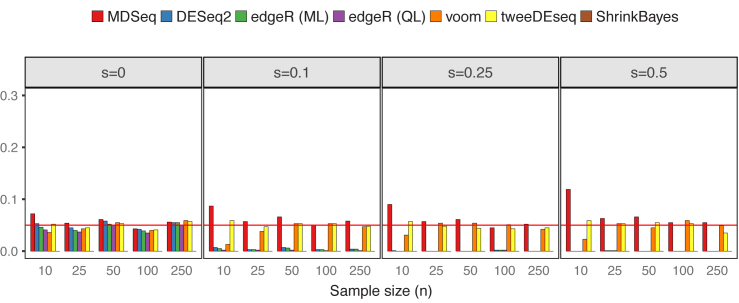
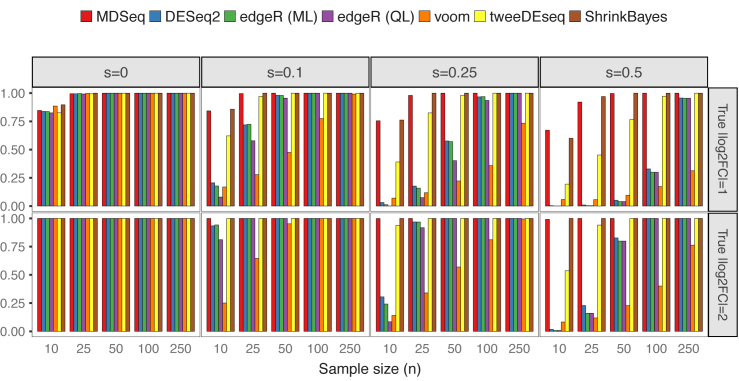
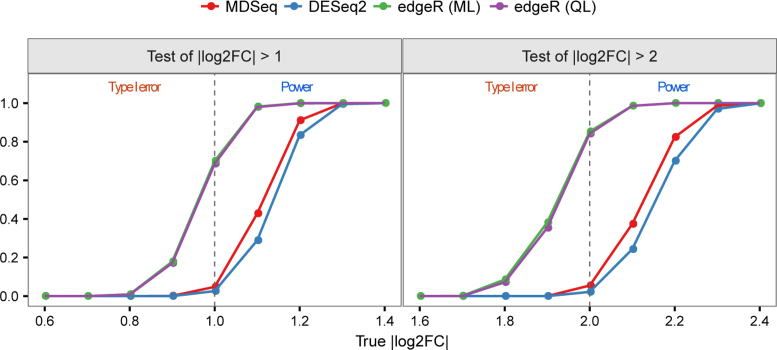
Rapidly decreasing cost of next-generation sequencing has led to the recent availability of large-scale RNA-seq data, that empowers the analysis of gene expression variability, in addition to gene expression means. In this paper, we present the MDSeq, based on the coefficient of dispersion, to provide robust and computationally efficient analysis of both gene expression means and variability on RNA-seq counts. The MDSeq utilizes a novel reparametrization of the negative binomial to provide flexible generalized linear models (GLMs) on both the mean and dispersion. We address challenges of analyzing large-scale RNA-seq data via several new developments to provide a comprehensive toolset that models technical excess zeros, identifies outliers efficiently, and evaluates differential expressions at biologically interesting levels. We evaluated performances of the MDSeq using simulated data when the ground truths are known. Results suggest that the MDSeq often outperforms current methods for the analysis of gene expression mean and variability. Moreover, the MDSeq is applied in two real RNA-seq studies, in which we identified functionally relevant genes and gene pathways. Specifically, the analysis of gene expression variability with the MDSeq on the GTEx human brain tissue data has identified pathways associated with common neurodegenerative disorders when gene expression means were conserved.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data.Genome Biol. 2013;14(9):R95. doi: 10.1186/gb-2013-14-9-r95. Genome Biol. 2013. PMID: 24020486 Free PMC article.
-
Detection of high variability in gene expression from single-cell RNA-seq profiling.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):508. doi: 10.1186/s12864-016-2897-6. BMC Genomics. 2016. PMID: 27556924 Free PMC article.
-
RNA-Seq Data Analysis in Galaxy.Methods Mol Biol. 2021;2284:367-392. doi: 10.1007/978-1-0716-1307-8_20. Methods Mol Biol. 2021. PMID: 33835453
-
Differential Expression Analysis of RNA-seq Reads: Overview, Taxonomy, and Tools.IEEE/ACM Trans Comput Biol Bioinform. 2020 Mar-Apr;17(2):566-586. doi: 10.1109/TCBB.2018.2873010. Epub 2018 Oct 1. IEEE/ACM Trans Comput Biol Bioinform. 2020. PMID: 30281477 Review.
-
Single-cell RNA-seq: advances and future challenges.Nucleic Acids Res. 2014 Aug;42(14):8845-60. doi: 10.1093/nar/gku555. Epub 2014 Jul 22. Nucleic Acids Res. 2014. PMID: 25053837 Free PMC article. Review.
Cited by
-
Developmental Programming: Prenatal Testosterone Excess on Liver and Muscle Coding and Noncoding RNA in Female Sheep.Endocrinology. 2022 Jan 1;163(1):bqab225. doi: 10.1210/endocr/bqab225. Endocrinology. 2022. PMID: 34718504 Free PMC article.
-
clrDV: a differential variability test for RNA-Seq data based on the skew-normal distribution.PeerJ. 2023 Sep 29;11:e16126. doi: 10.7717/peerj.16126. eCollection 2023. PeerJ. 2023. PMID: 37790621 Free PMC article.
-
Robust and Adaptive Non-Parametric Tests for Detecting General Distributional Shifts in Gene Expression.bioRxiv [Preprint]. 2025 Mar 11:2025.03.06.641952. doi: 10.1101/2025.03.06.641952. bioRxiv. 2025. PMID: 40161649 Free PMC article. Preprint.
-
Coordinated analysis of exon and intron data reveals novel differential gene expression changes.Sci Rep. 2020 Sep 24;10(1):15669. doi: 10.1038/s41598-020-72482-w. Sci Rep. 2020. PMID: 32973253 Free PMC article.
-
Detection of genes with differential expression dispersion unravels the role of autophagy in cancer progression.PLoS Comput Biol. 2023 Mar 9;19(3):e1010342. doi: 10.1371/journal.pcbi.1010342. eCollection 2023 Mar. PLoS Comput Biol. 2023. PMID: 36893104 Free PMC article.
References
-
- Markert J.M., Fuller C.M., Gillespie G.Y., Bubien J.K., McLean L.A., Hong R.L., Lee K., Gullans S.R., Mapstone T.B., Benos D.J.. Differential gene expression profiling in human brain tumors. Physiol. Genomics. 2001; 5:21–33. - PubMed
-
- Jiang Y., Harlocker S.L., Molesh D.A., Dillon D.C., Stolk J.A., Houghton R.L., Repasky E.A., Badaro R., Reed S.G., Xu J.. Discovery of differentially expressed genes in human breast cancer using subtracted cDNA libraries and cDNA microarrays. Oncogene. 2002; 21:2270–2282. - PubMed
-
- Richer J.K., Jacobsen B.M., Manning N.G., Abel M.G., Wolf D.M., Horwitz K.B.. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J. Biol. Chem. 2002; 277:5209–5218. - PubMed
-
- Howell B.G., Solish N., Lu C., Watanabe H., Mamelak A.J., Freed I., Wang B., Sauder D.N.. Microarray profiles of human basal cell carcinoma: insights into tumor growth and behavior. J. Dermatol. Sci. 2005; 39:39–51. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
