

Bioinformatic analysis of bacteria and host cell dual RNA-sequencing experiments
- PMID: 28535295
- PMCID: PMC6291798
- DOI: 10.1093/bib/bbx043
Bioinformatic analysis of bacteria and host cell dual RNA-sequencing experiments
Abstract
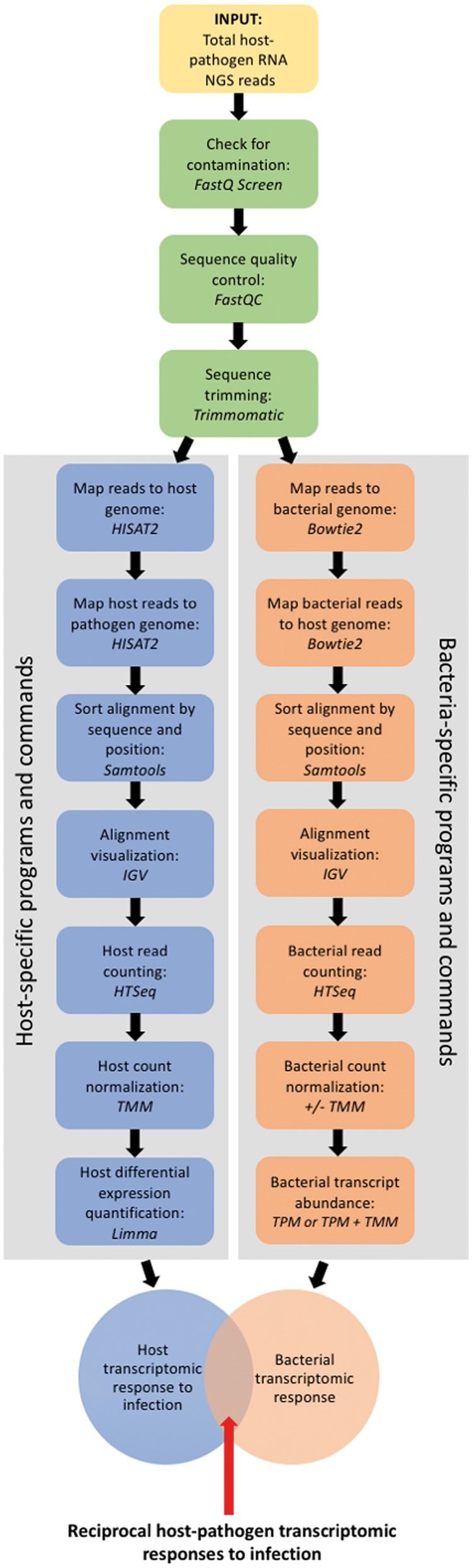
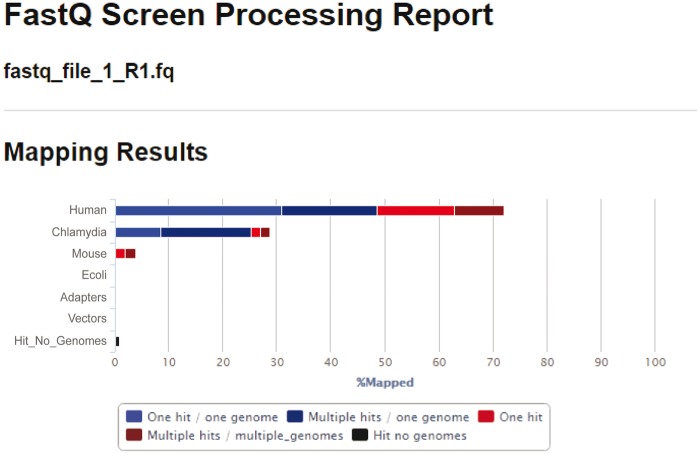
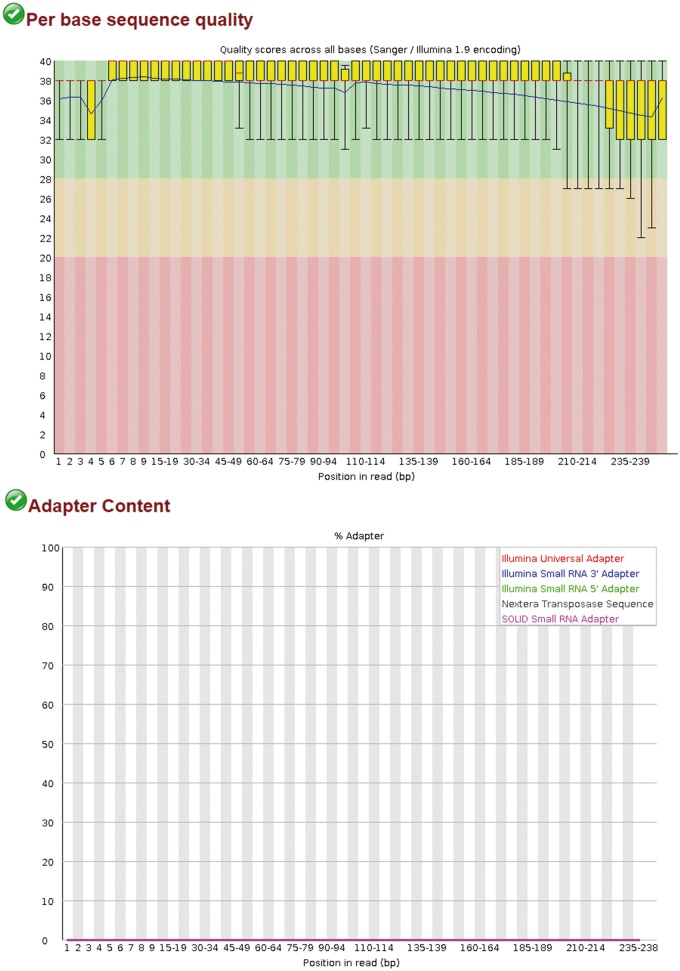
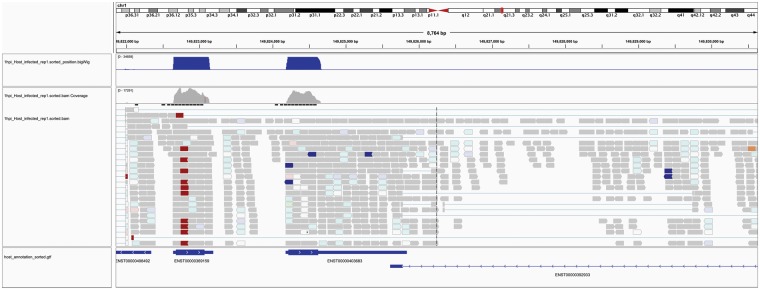
Bacterial pathogens subvert host cells by manipulating cellular pathways for survival and replication; in turn, host cells respond to the invading pathogen through cascading changes in gene expression. Deciphering these complex temporal and spatial dynamics to identify novel bacterial virulence factors or host response pathways is crucial for improved diagnostics and therapeutics. Dual RNA sequencing (dRNA-Seq) has recently been developed to simultaneously capture host and bacterial transcriptomes from an infected cell. This approach builds on the high sensitivity and resolution of RNA sequencing technology and is applicable to any bacteria that interact with eukaryotic cells, encompassing parasitic, commensal or mutualistic lifestyles. Several laboratory protocols have been presented that outline the collection, extraction and sequencing of total RNA for dRNA-Seq experiments, but there is relatively little guidance available for the detailed bioinformatic analyses required. This protocol outlines a typical dRNA-Seq experiment, based on a Chlamydia trachomatis-infected host cell, with a detailed description of the necessary bioinformatic analyses with currently available software tools.
Figures
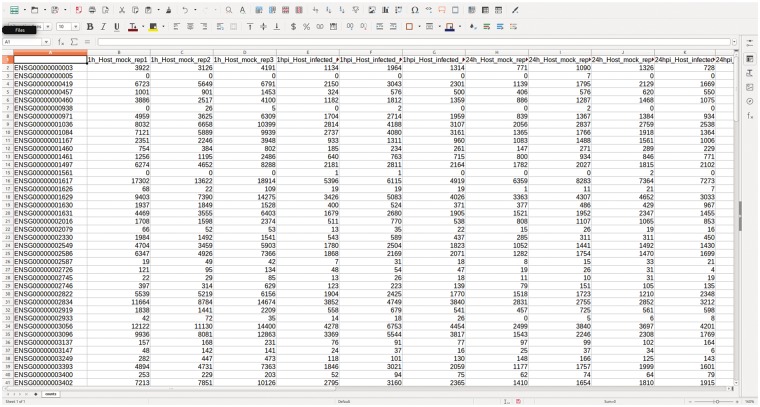
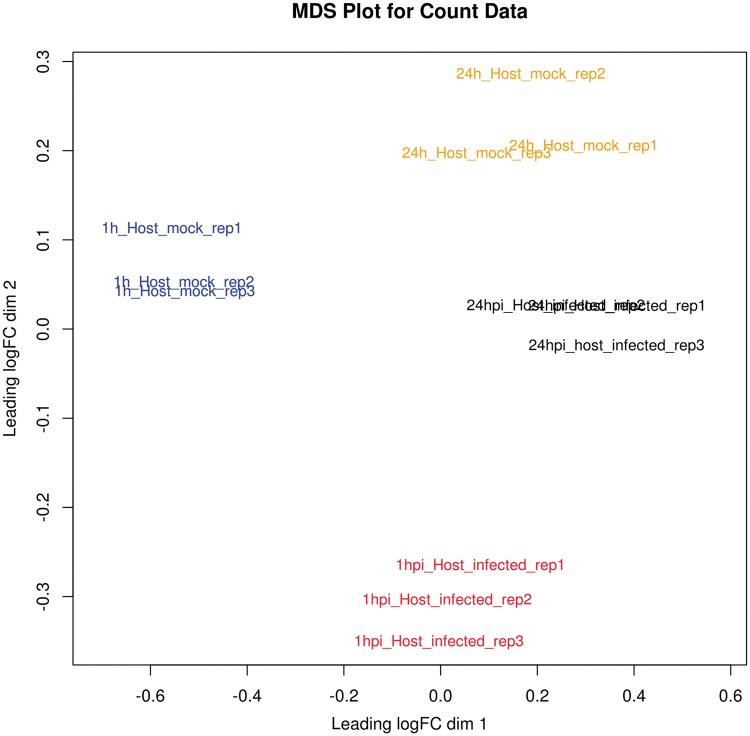
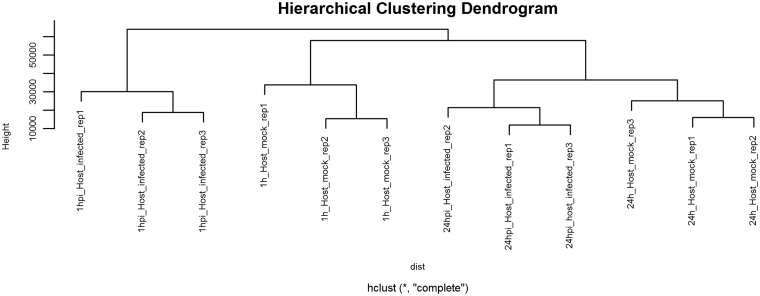
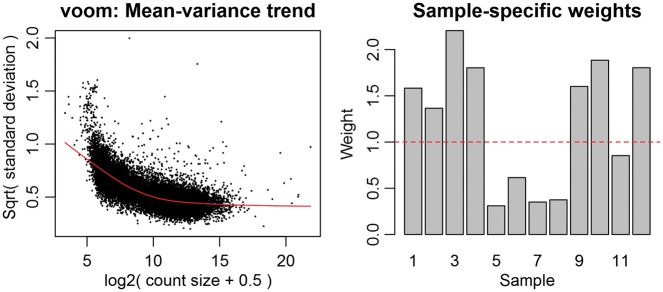
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
