

MetaSRA: normalized human sample-specific metadata for the Sequence Read Archive
- PMID: 28535296
- PMCID: PMC5870770
- DOI: 10.1093/bioinformatics/btx334
MetaSRA: normalized human sample-specific metadata for the Sequence Read Archive
Abstract
Motivation: The NCBI's Sequence Read Archive (SRA) promises great biological insight if one could analyze the data in the aggregate; however, the data remain largely underutilized, in part, due to the poor structure of the metadata associated with each sample. The rules governing submissions to the SRA do not dictate a standardized set of terms that should be used to describe the biological samples from which the sequencing data are derived. As a result, the metadata include many synonyms, spelling variants and references to outside sources of information. Furthermore, manual annotation of the data remains intractable due to the large number of samples in the archive. For these reasons, it has been difficult to perform large-scale analyses that study the relationships between biomolecular processes and phenotype across diverse diseases, tissues and cell types present in the SRA.
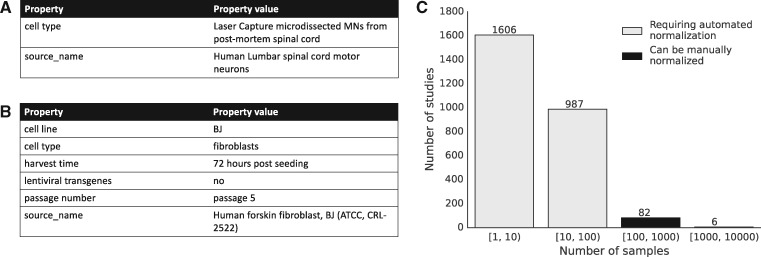
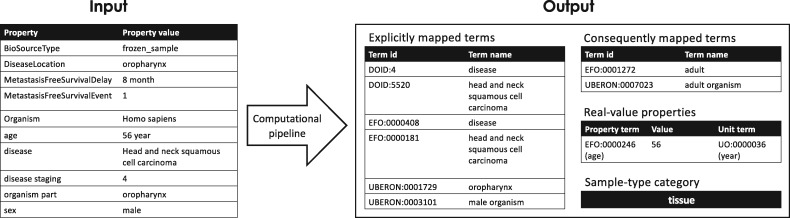
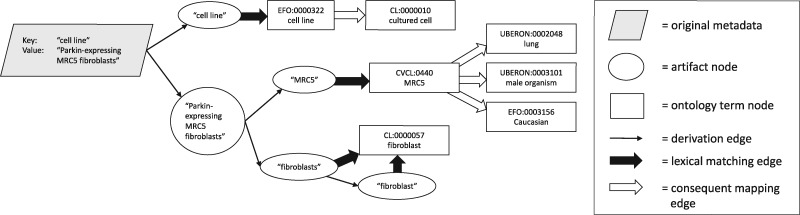
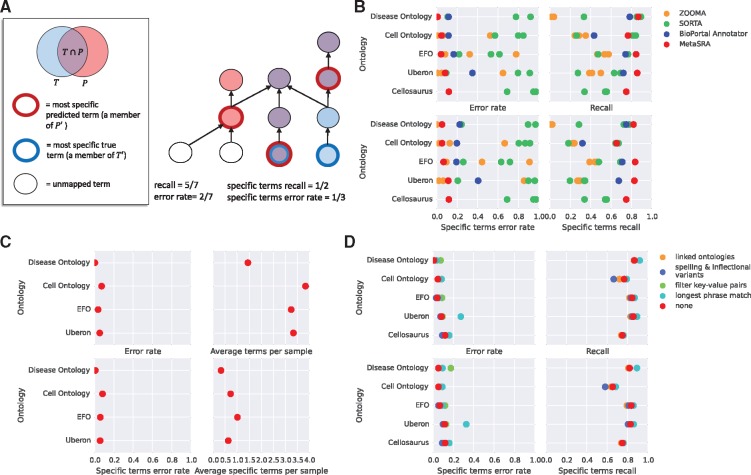
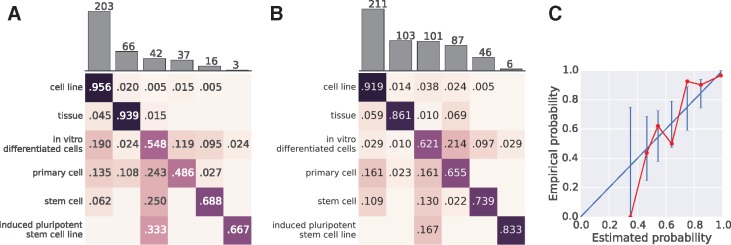
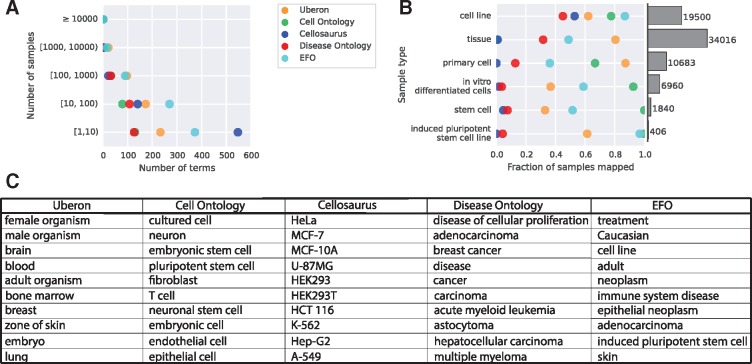
Results: We present MetaSRA, a database of normalized SRA human sample-specific metadata following a schema inspired by the metadata organization of the ENCODE project. This schema involves mapping samples to terms in biomedical ontologies, labeling each sample with a sample-type category, and extracting real-valued properties. We automated these tasks via a novel computational pipeline.
Availability and implementation: The MetaSRA is available at metasra.biostat.wisc.edu via both a searchable web interface and bulk downloads. Software implementing our computational pipeline is available at http://github.com/deweylab/metasra-pipeline.
Contact: cdewey@biostat.wisc.edu.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2017. Published by Oxford University Press.
Figures
References
-
- Bartolini I. et al. (2002). String matching with metric trees using an approximate distance In: Proceedings of the 9th International Symposium on String Processing and Information Retrieval, SPIRE 2002, Springer-Verlag, London, UK, pp. 271–283.
-
- Bröcker J., Smith L. (2007) Increasing the reliability of reliability diagrams. Weather Forecasting, 22, 651–661.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
