

Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models
- PMID: 28535794
- PMCID: PMC5442865
- DOI: 10.1186/s13058-017-0851-4
Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models
Abstract
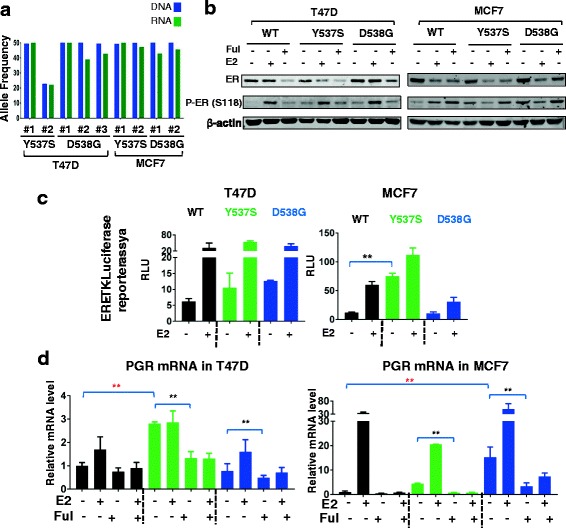
Background: Mutations in the estrogen receptor alpha (ERα) 1 gene (ESR1) are frequently detected in ER+ metastatic breast cancer, and there is increasing evidence that these mutations confer endocrine resistance in breast cancer patients with advanced disease. However, their functional role is not well-understood, at least in part due to a lack of ESR1 mutant models. Here, we describe the generation and characterization of genome-edited T47D and MCF7 breast cancer cell lines with the two most common ESR1 mutations, Y537S and D538G.
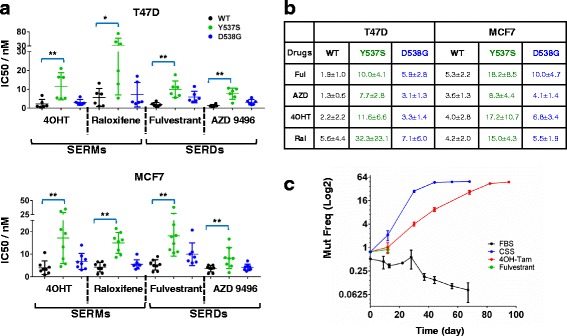
Methods: Genome editing was performed using CRISPR and adeno-associated virus (AAV) technologies to knock-in ESR1 mutations into T47D and MCF7 cell lines, respectively. Various techniques were utilized to assess the activity of mutant ER, including transactivation, growth and chromatin-immunoprecipitation (ChIP) assays. The level of endocrine resistance was tested in mutant cells using a number of selective estrogen receptor modulators (SERMs) and degraders (SERDs). RNA sequencing (RNA-seq) was employed to study gene targets of mutant ER.
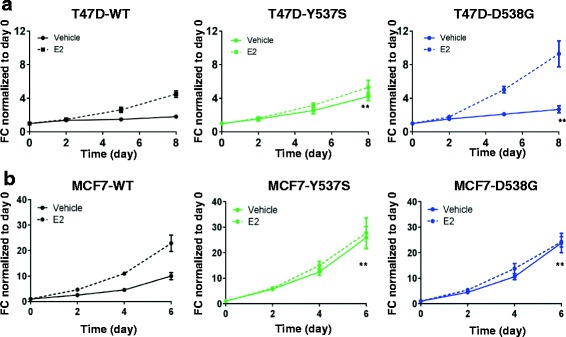
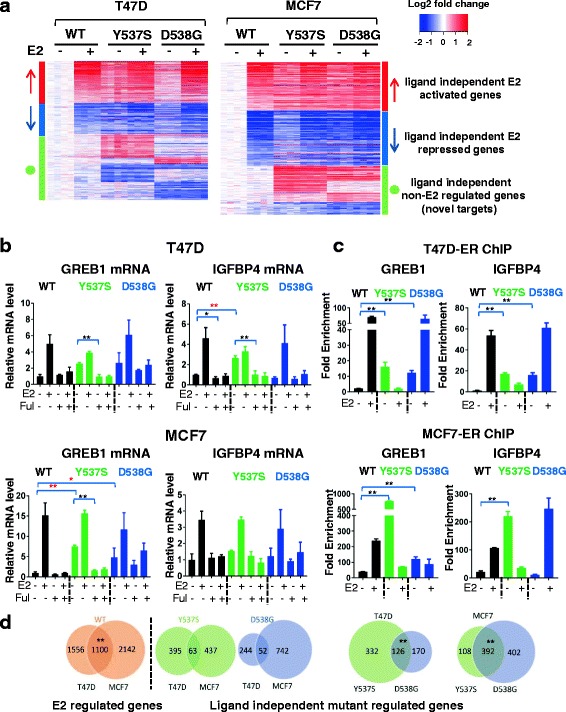
Results: Cells with ESR1 mutations displayed ligand-independent ER activity, and were resistant to several SERMs and SERDs, with cell line and mutation-specific differences with respect to magnitude of effect. The SERD AZ9496 showed increased efficacy compared to other drugs tested. Wild-type and mutant cell co-cultures demonstrated a unique evolution of mutant cells under estrogen deprivation and tamoxifen treatment. Transcriptome analysis confirmed ligand-independent regulation of ERα target genes by mutant ERα, but also identified novel target genes, some of which are involved in metastasis-associated phenotypes. Despite significant overlap in the ligand-independent genes between Y537S and D538G, the number of mutant ERα-target genes shared between the two cell lines was limited, suggesting context-dependent activity of the mutant receptor. Some genes and phenotypes were unique to one mutation within a given cell line, suggesting a mutation-specific effect.
Conclusions: Taken together, ESR1 mutations in genome-edited breast cancer cell lines confer ligand-independent growth and endocrine resistance. These biologically relevant models can be used for further mechanistic and translational studies, including context-specific and mutation site-specific analysis of the ESR1 mutations.
Keywords: ESR1 mutations; Endocrine resistance; Genome-edited cells; Metastatic breast cancer; RNA-seq.
Figures
Similar articles
-
Upregulation of IRS1 Enhances IGF1 Response in Y537S and D538G ESR1 Mutant Breast Cancer Cells.Endocrinology. 2018 Jan 1;159(1):285-296. doi: 10.1210/en.2017-00693. Endocrinology. 2018. PMID: 29029116 Free PMC article.
-
Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer.Clin Cancer Res. 2015 Nov 15;21(22):5121-5130. doi: 10.1158/1078-0432.CCR-15-0360. Epub 2015 May 19. Clin Cancer Res. 2015. PMID: 25991817 Free PMC article.
-
Stereospecific lasofoxifene derivatives reveal the interplay between estrogen receptor alpha stability and antagonistic activity in ESR1 mutant breast cancer cells.Elife. 2022 May 16;11:e72512. doi: 10.7554/eLife.72512. Elife. 2022. PMID: 35575456 Free PMC article.
-
The association between type of endocrine therapy and development of estrogen receptor-1 mutation(s) in patients with hormone-sensitive advanced breast cancer: A systematic review and meta-analysis of randomized and non-randomized trials.Biochim Biophys Acta Rev Cancer. 2019 Dec;1872(2):188315. doi: 10.1016/j.bbcan.2019.188315. Epub 2019 Oct 21. Biochim Biophys Acta Rev Cancer. 2019. PMID: 31647985
-
Strategies to degrade estrogen receptor α in primary and ESR1 mutant-expressing metastatic breast cancer.Mol Cell Endocrinol. 2019 Jan 15;480:107-121. doi: 10.1016/j.mce.2018.10.020. Epub 2018 Oct 31. Mol Cell Endocrinol. 2019. PMID: 30389467 Review.
Cited by
-
Estrogen Receptor Alpha and ESR1 Mutations in Breast Cancer.Adv Exp Med Biol. 2022;1390:171-194. doi: 10.1007/978-3-031-11836-4_10. Adv Exp Med Biol. 2022. PMID: 36107319
-
Preexisting Somatic Mutations of Estrogen Receptor Alpha (ESR1) in Early-Stage Primary Breast Cancer.JNCI Cancer Spectr. 2021 Apr 22;5(2):pkab028. doi: 10.1093/jncics/pkab028. eCollection 2021 Apr. JNCI Cancer Spectr. 2021. PMID: 33937624 Free PMC article.
-
EstroGene database reveals diverse temporal, context-dependent and directional estrogen receptor regulomes in breast cancer.bioRxiv [Preprint]. 2023 Feb 2:2023.01.30.526388. doi: 10.1101/2023.01.30.526388. bioRxiv. 2023. Update in: Cancer Res. 2023 Aug 15;83(16):2656-2674. doi: 10.1158/0008-5472.CAN-23-0539. PMID: 36778377 Free PMC article. Updated. Preprint.
-
Antagonists for Constitutively Active Mutant Estrogen Receptors: Insights into the Roles of Antiestrogen-Core and Side-Chain.ACS Chem Biol. 2018 Dec 21;13(12):3374-3384. doi: 10.1021/acschembio.8b00877. Epub 2018 Nov 26. ACS Chem Biol. 2018. PMID: 30404440 Free PMC article.
-
CRISPR/Cas9 system in breast cancer therapy: advancement, limitations and future scope.Cancer Cell Int. 2022 Jul 25;22(1):234. doi: 10.1186/s12935-022-02654-3. Cancer Cell Int. 2022. PMID: 35879772 Free PMC article. Review.
References
-
- Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757–67. doi: 10.1158/1078-0432.CCR-13-2332. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
