

Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture
- PMID: 28536263
- PMCID: PMC5519369
- DOI: 10.1074/jbc.M117.784702
Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture
Abstract
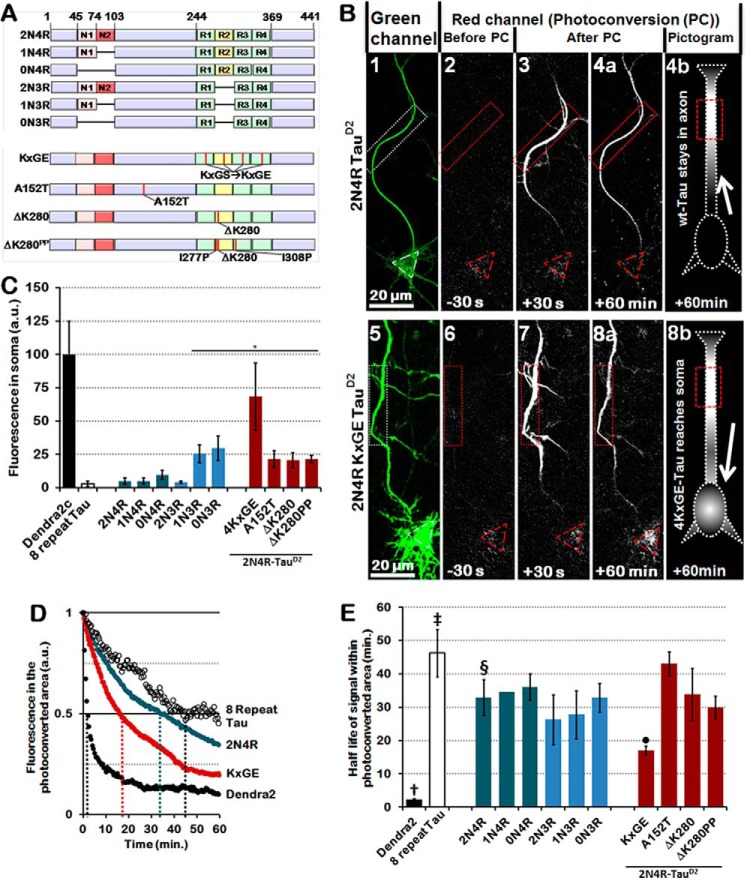
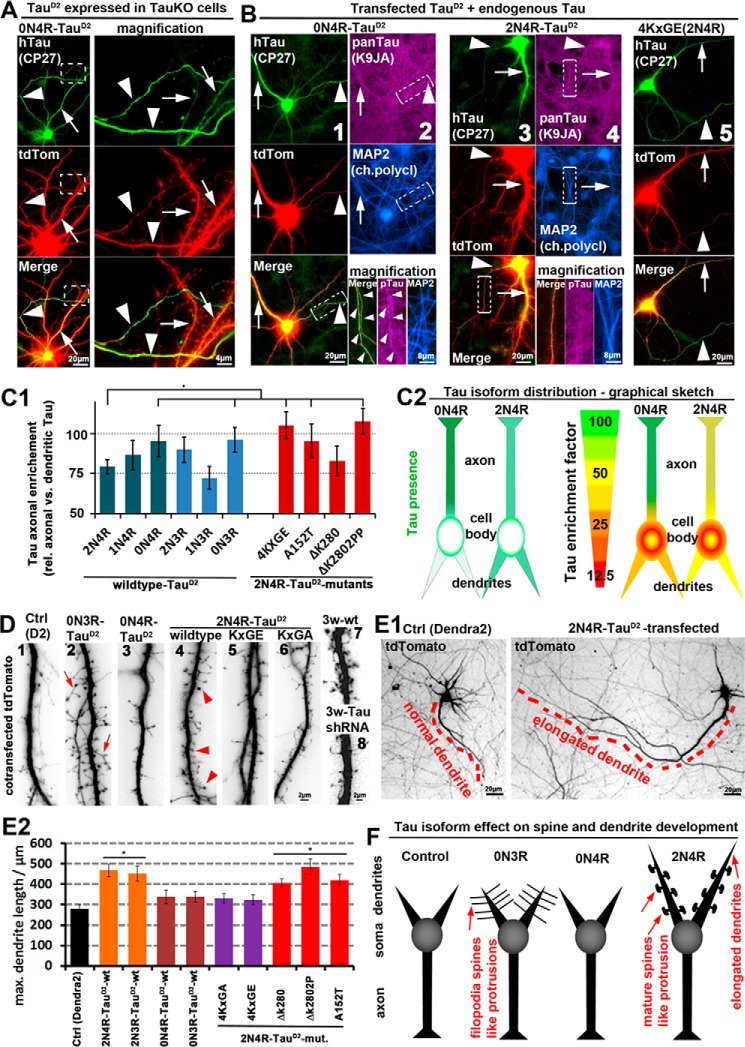
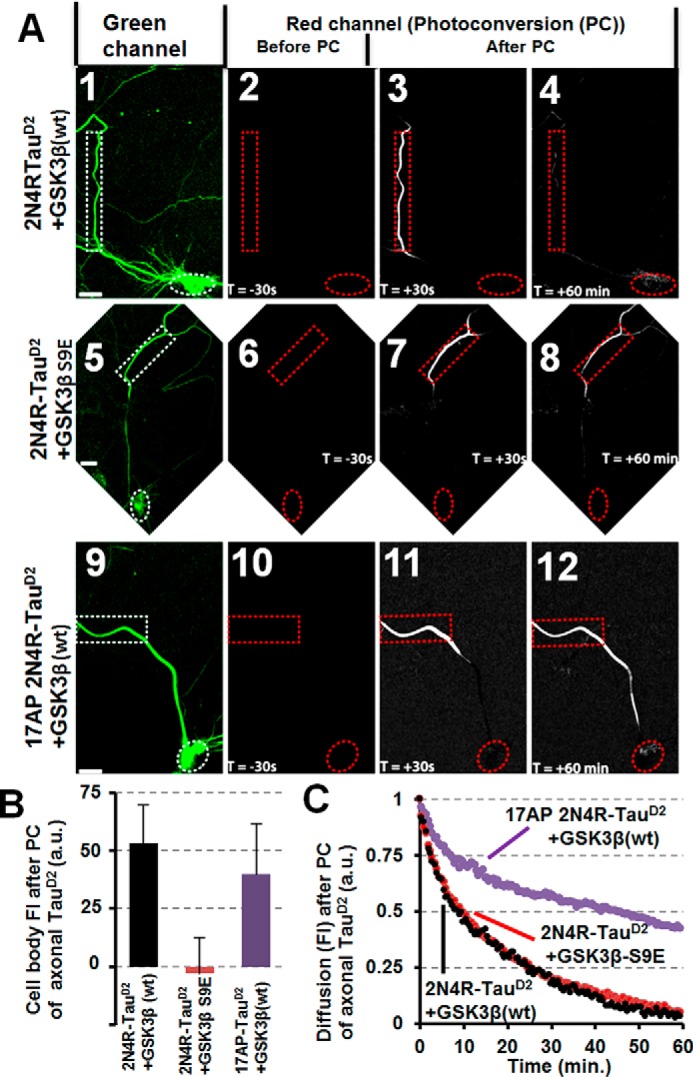
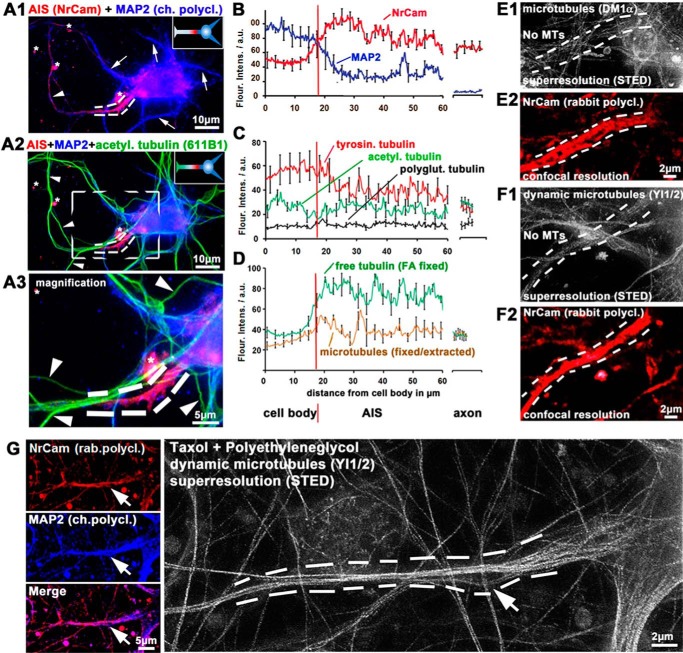
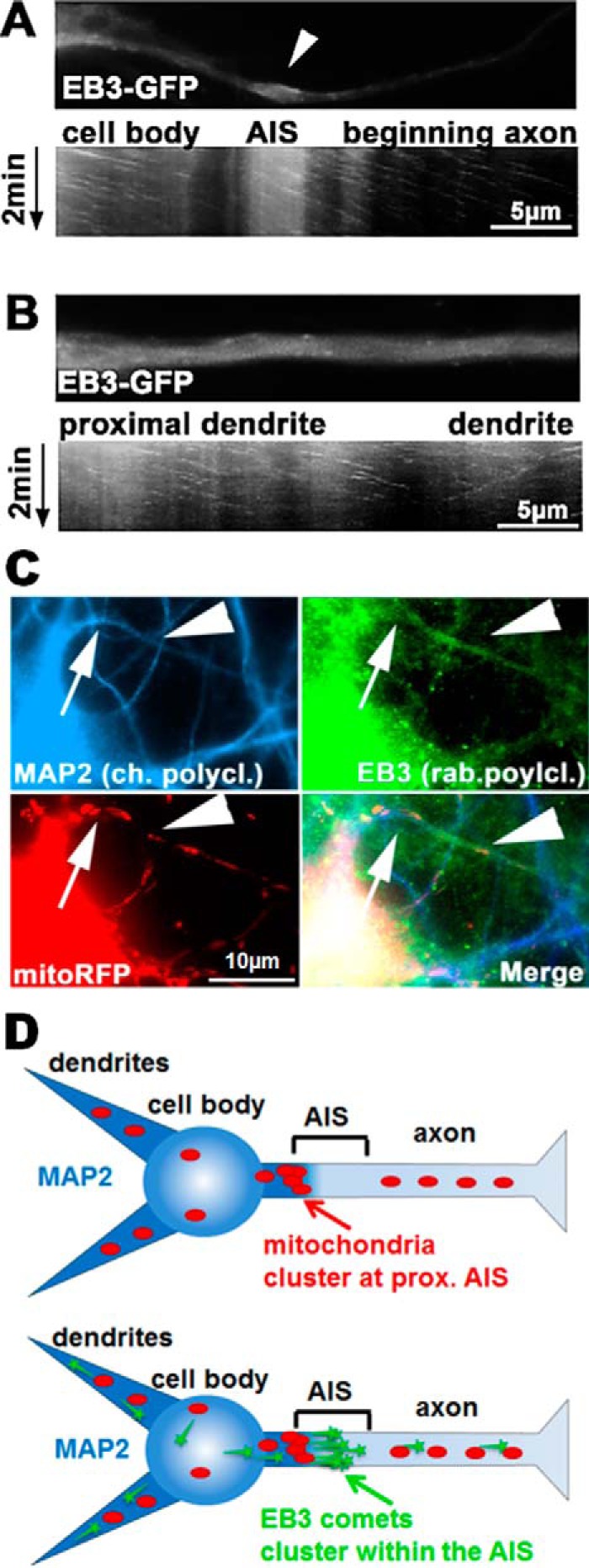
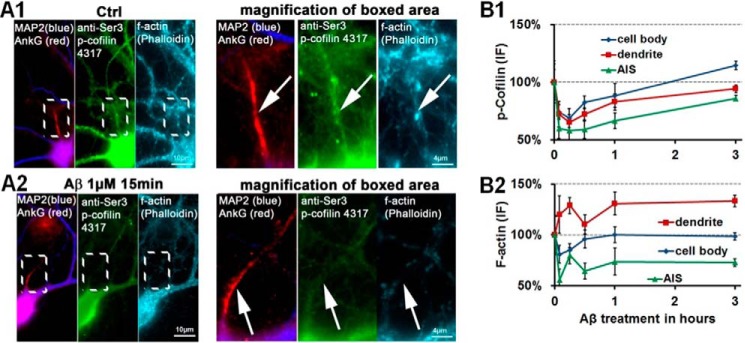
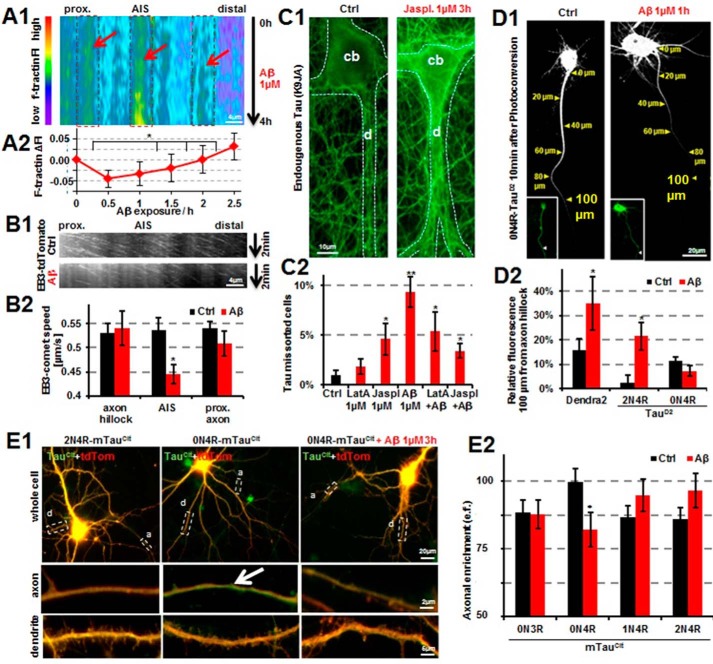
Subcellular mislocalization of the microtubule-associated protein Tau is a hallmark of Alzheimer disease (AD) and other tauopathies. Six Tau isoforms, differentiated by the presence or absence of a second repeat or of N-terminal inserts, exist in the human CNS, but their physiological and pathological differences have long remained elusive. Here, we investigated the properties and distributions of human and rodent Tau isoforms in primary forebrain rodent neurons. We found that the Tau diffusion barrier (TDB), located within the axon initial segment (AIS), controls retrograde (axon-to-soma) and anterograde (soma-to-axon) traffic of Tau. Tau isoforms without the N-terminal inserts were sorted efficiently into the axon. However, the longest isoform (2N4R-Tau) was partially retained in cell bodies and dendrites, where it accelerated spine and dendrite growth. The TDB (located within the AIS) was impaired when AIS components (ankyrin G, EB1) were knocked down or when glycogen synthase kinase-3β (GSK3β; an AD-associated kinase tethered to the AIS) was overexpressed. Using superresolution nanoscopy and live-cell imaging, we observed that microtubules within the AIS appeared highly dynamic, a feature essential for the TDB. Pathomechanistically, amyloid-β insult caused cofilin activation and F-actin remodeling and decreased microtubule dynamics in the AIS. Concomitantly with these amyloid-β-induced disruptions, the AIS/TDB sorting function failed, causing AD-like Tau missorting. In summary, we provide evidence that the human and rodent Tau isoforms differ in axodendritic sorting and amyloid-β-induced missorting and that the axodendritic distribution of Tau depends on AIS integrity.
Keywords: AIS; Alzheimer disease; Tau protein (Tau); amyloid-beta (Aβ); cell polarity; microtubule; neurodegeneration; tauopathy.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Wang Y., and Mandelkow E. (2016) Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21 - PubMed
-
- Goedert M., Spillantini M. G., Potier M. C., Ulrich J., and Crowther R. A. (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8, 393–399 - PMC - PubMed
-
- Trabzuni D., Wray S., Vandrovcova J., Ramasamy A., Walker R., Smith C., Luk C., Gibbs J. R., Dillman A., Hernandez D. G., Arepalli S., Singleton A. B., Cookson M. R., Pittman A. M., de Silva R., et al. (2012) MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum. Mol. Genet. 21, 4094–4103 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
