

Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease
- PMID: 28536304
- PMCID: PMC5494504
- DOI: 10.15252/emmm.201707608
Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease
Abstract
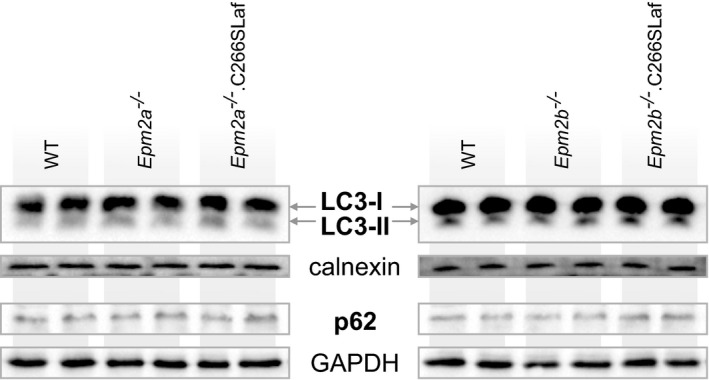
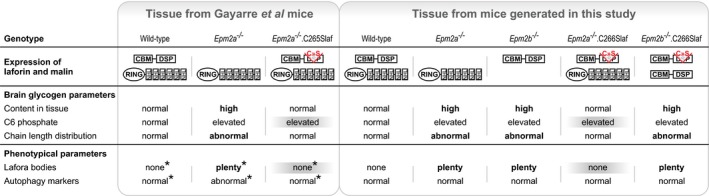
Lafora disease (LD) is a fatal progressive epilepsy essentially caused by loss-of-function mutations in the glycogen phosphatase laforin or the ubiquitin E3 ligase malin. Glycogen in LD is hyperphosphorylated and poorly hydrosoluble. It precipitates and accumulates into neurotoxic Lafora bodies (LBs). The leading LD hypothesis that hyperphosphorylation causes the insolubility was recently challenged by the observation that phosphatase-inactive laforin rescues the laforin-deficient LD mouse model, apparently through correction of a general autophagy impairment. We were for the first time able to quantify brain glycogen phosphate. We also measured glycogen content and chain lengths, LBs, and autophagy markers in several laforin- or malin-deficient mouse lines expressing phosphatase-inactive laforin. We find that: (i) in laforin-deficient mice, phosphatase-inactive laforin corrects glycogen chain lengths, and not hyperphosphorylation, which leads to correction of glycogen amounts and prevention of LBs; (ii) in malin-deficient mice, phosphatase-inactive laforin confers no correction; (iii) general impairment of autophagy is not necessary in LD We conclude that laforin's principle function is to control glycogen chain lengths, in a malin-dependent fashion, and that loss of this control underlies LD.
Keywords: Lafora disease; glycogen chain length; glycogen phosphorylation; laforin; malin.
© 2017 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
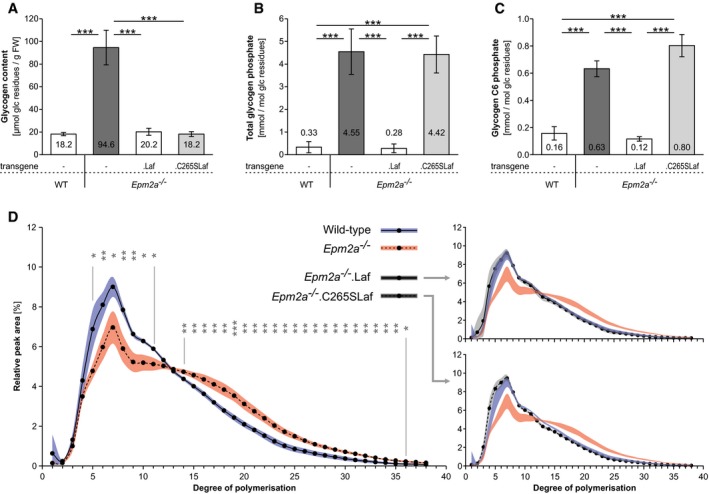
Glycogen content.
Glycogen total phosphate content.
Glycogen carbon C6 phosphate content.
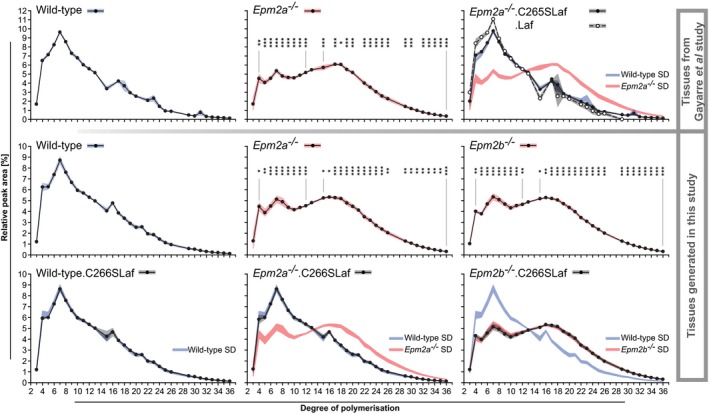
Chain length distribution analyses.
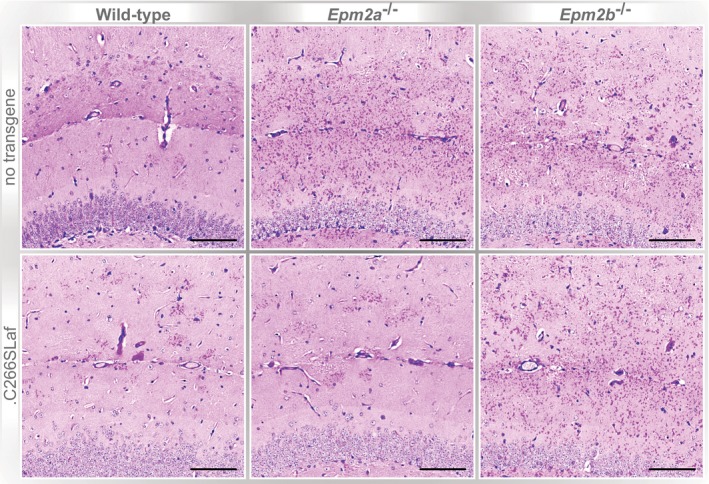
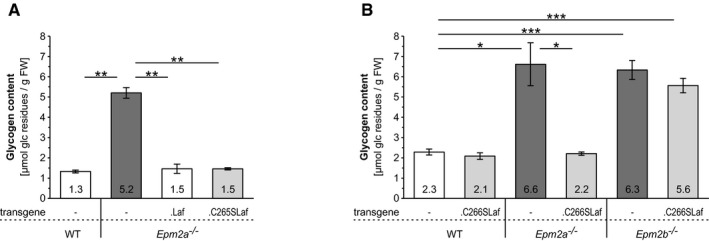
Brain glycogen levels in the presence or absence of WT (.Laf) or C265S mutated (.C265Laf) murine laforin transgene (tissues from the Gayarre et al, 2014 study).
Brain glycogen levels of WT, Epm2a −/− and Epm2b −/− mice in the presence or absence of human C266S mutated laforin (.C266SLaf) (tissues from the new mice generated in the present study).
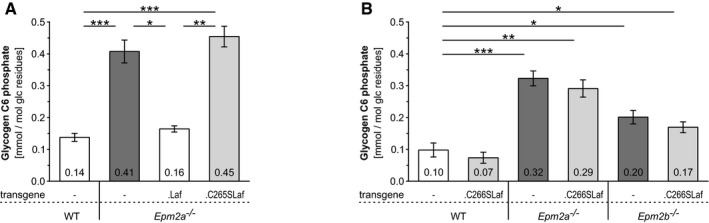
Brain glycogen carbon C6 phosphate levels in the presence or absence of WT (.Laf) or C265S mutated (.C265Laf) murine laforin transgene.
Brain glycogen C6 phosphate levels of WT, Epm2a −/− and Epm2b −/− mice in the presence or absence of human C266S mutated laforin (.C266SLaf).
References
-
- Cenci U, Nitschke F, Steup M, Minassian BA, Colleoni C, Ball SG (2014) Transition from glycogen to starch metabolism in Archaeplastida . Trends Plant Sci 19: 18–28 - PubMed
-
- Chan EM, Ackerley CA, Lohi H, Ianzano L, Cortez MA, Shannon P, Scherer SW, Minassian BA (2004) Laforin preferentially binds the neurotoxic starch‐like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum Mol Genet 13: 1117–1129 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
