

Comparative Proteomic Analysis of Mycobacterium tuberculosis Lineage 7 and Lineage 4 Strains Reveals Differentially Abundant Proteins Linked to Slow Growth and Virulence
- PMID: 28536560
- PMCID: PMC5423352
- DOI: 10.3389/fmicb.2017.00795
Comparative Proteomic Analysis of Mycobacterium tuberculosis Lineage 7 and Lineage 4 Strains Reveals Differentially Abundant Proteins Linked to Slow Growth and Virulence
Abstract
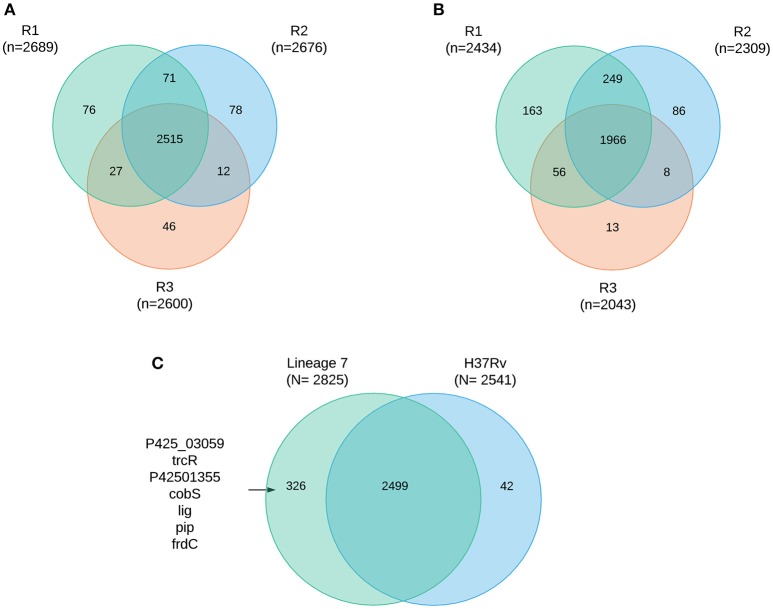
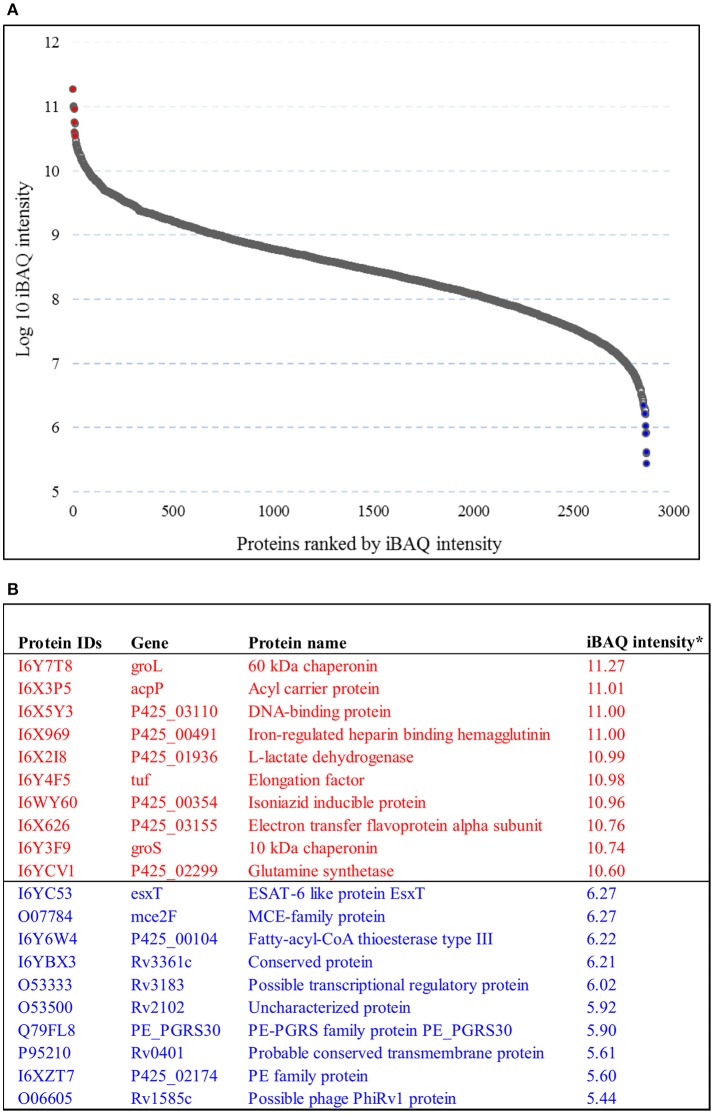
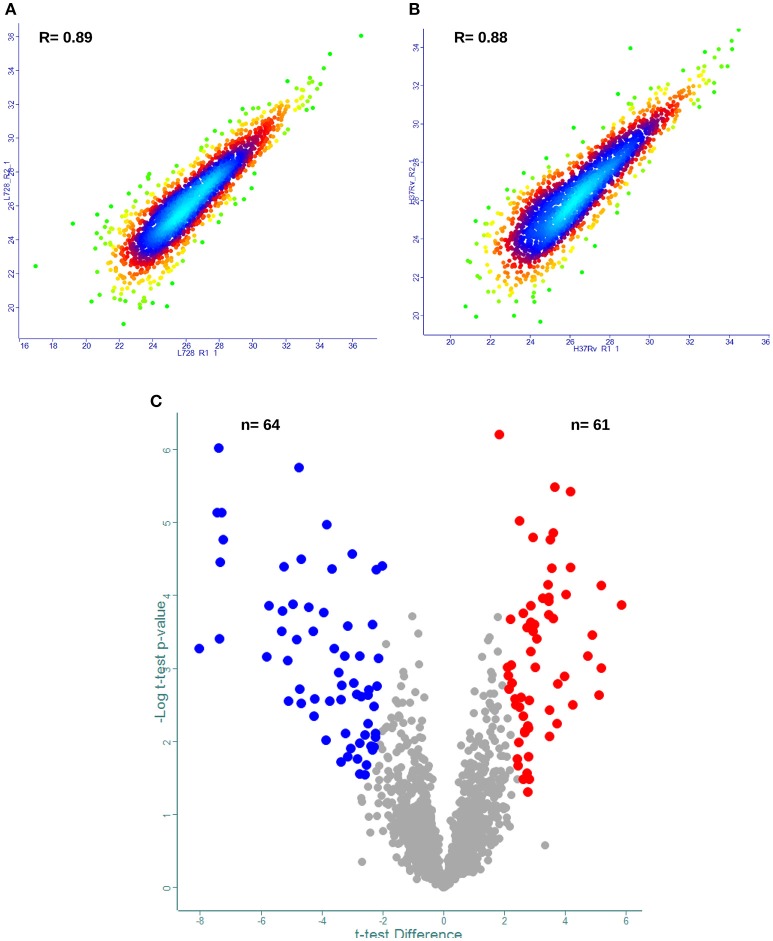
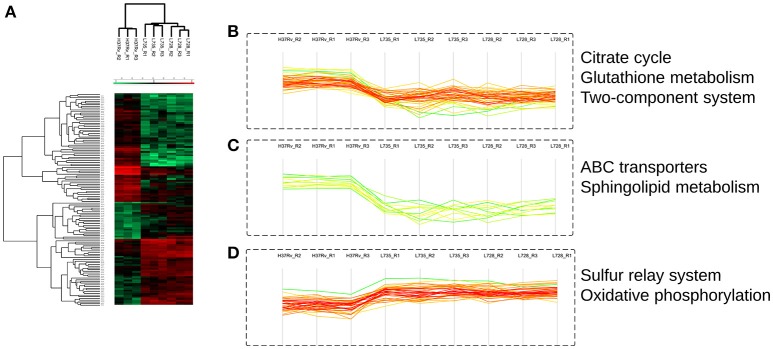
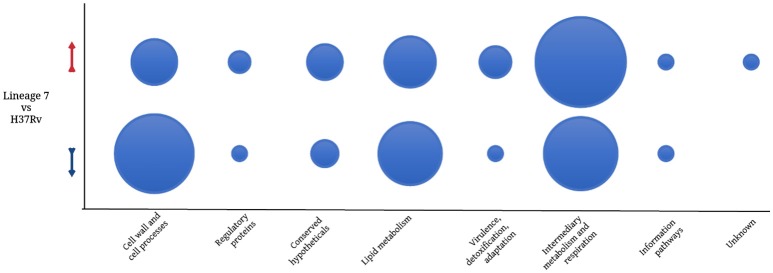
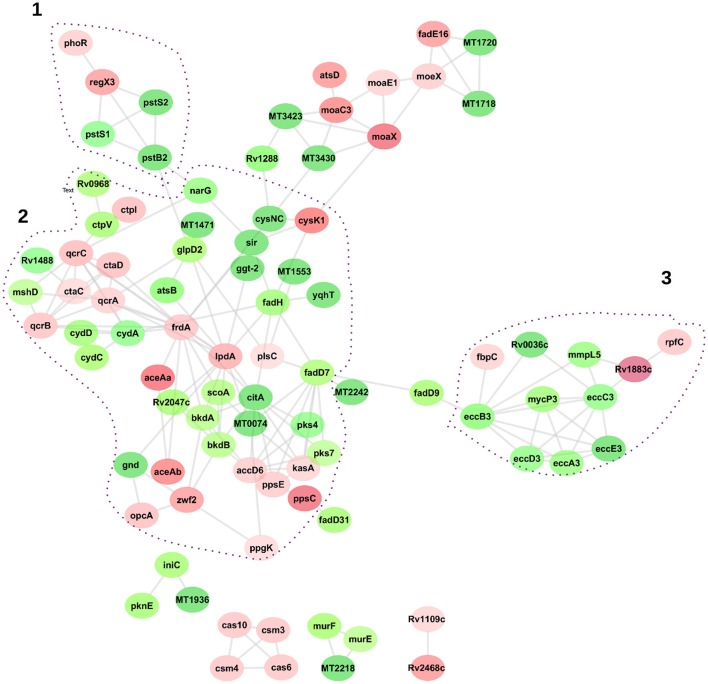
In order to decipher the nature of the slowly growing Mycobacterium tuberculosis (M.tuberculosis) lineage 7, the differentially abundant proteins in strains of M. tuberculosis lineage 7 and lineage 4 were defined. Comparative proteomic analysis by mass spectrometry was employed to identify, quantitate and compare the protein profiles of strains from the two M. tuberculosis lineages. Label-free peptide quantification of whole cells from M. tuberculosis lineage 7 and 4 yielded the identification of 2825 and 2541 proteins, respectively. A combined total of 2867 protein groups covering 71% of the predicted M. tuberculosis proteome were identified. The abundance of 125 proteins in M. tuberculosis lineage 7 and 4 strains was significantly altered. Notably, the analysis showed that a number of M. tuberculosis proteins involved in growth and virulence were less abundant in lineage 7 strains compared to lineage 4. Five ABC transporter proteins, three phosphate binding proteins essential for inorganic phosphate uptake, and six components of the type 7 secretion system ESX-3 involved in iron acquisition were less abundant in M. tuberculosis lineage 7. This proteogenomic analysis provided an insight into the lineage 7-specific protein profile which may provide clues to understanding the differential properties of lineage 7 strains in terms of slow growth, survival fitness, and pathogenesis.
Keywords: Ethiopia; Mycobacterium tuberculosis; lineage 7; mass spectrometry; proteomics; tuberculosis; type 7 secretion.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources