

NRAS mutations in cutaneous T cell lymphoma (CTCL) sensitize tumors towards treatment with the multikinase inhibitor Sorafenib
- PMID: 28537899
- PMCID: PMC5542218
- DOI: 10.18632/oncotarget.17669
NRAS mutations in cutaneous T cell lymphoma (CTCL) sensitize tumors towards treatment with the multikinase inhibitor Sorafenib
Abstract
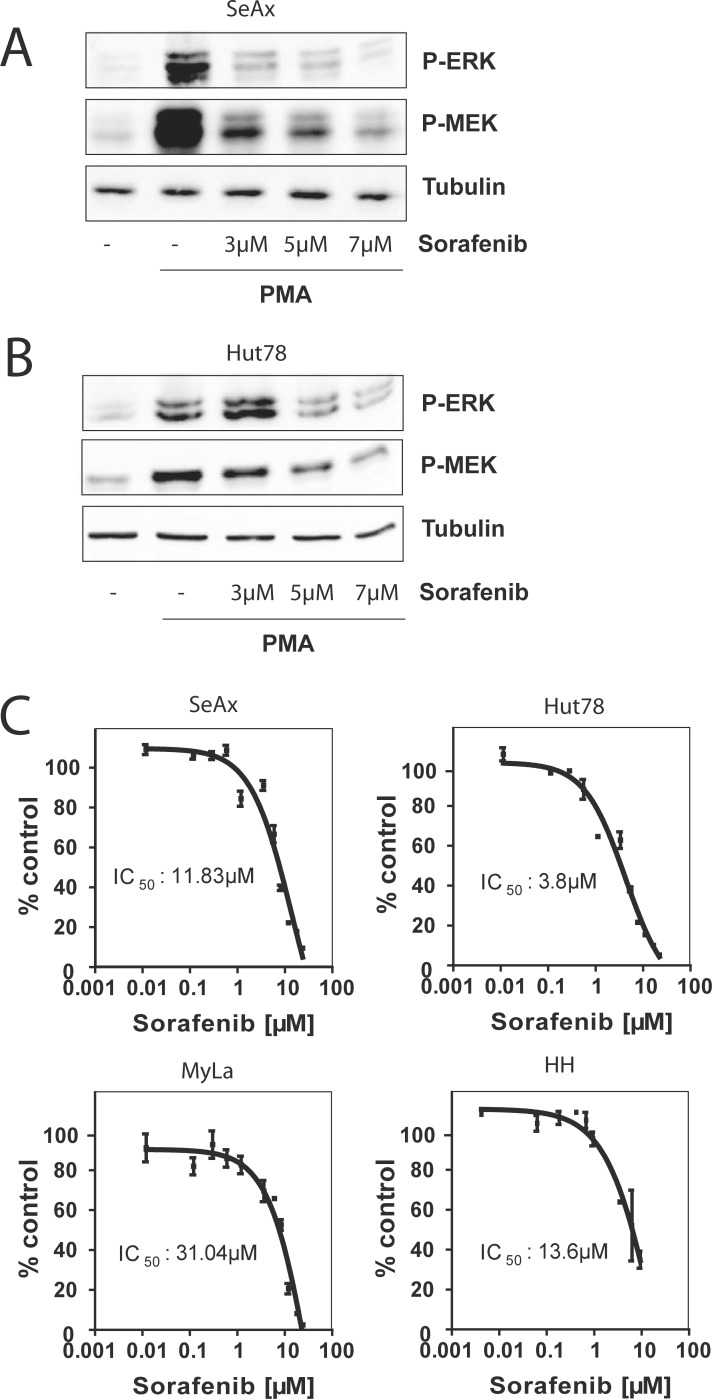
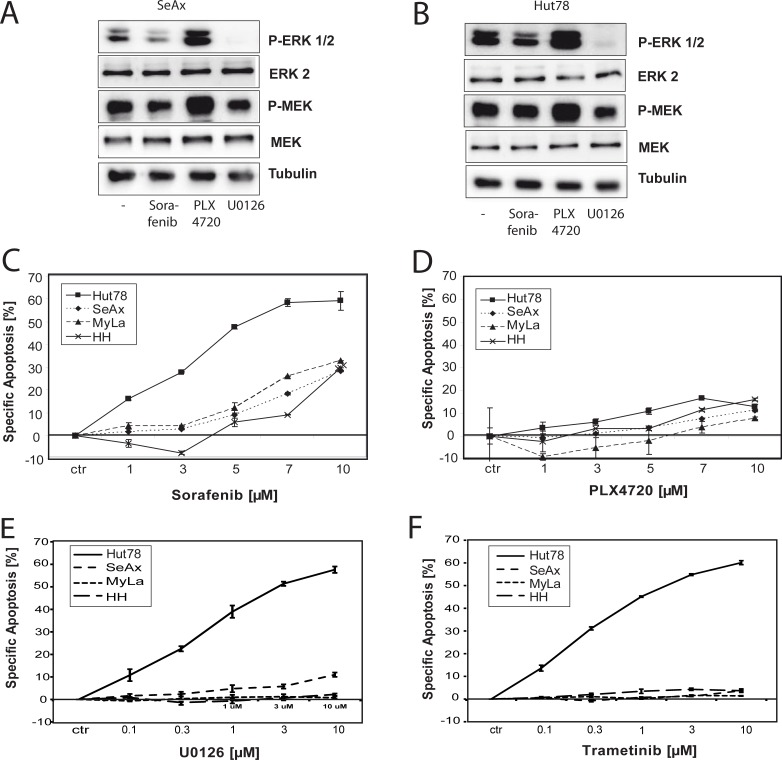
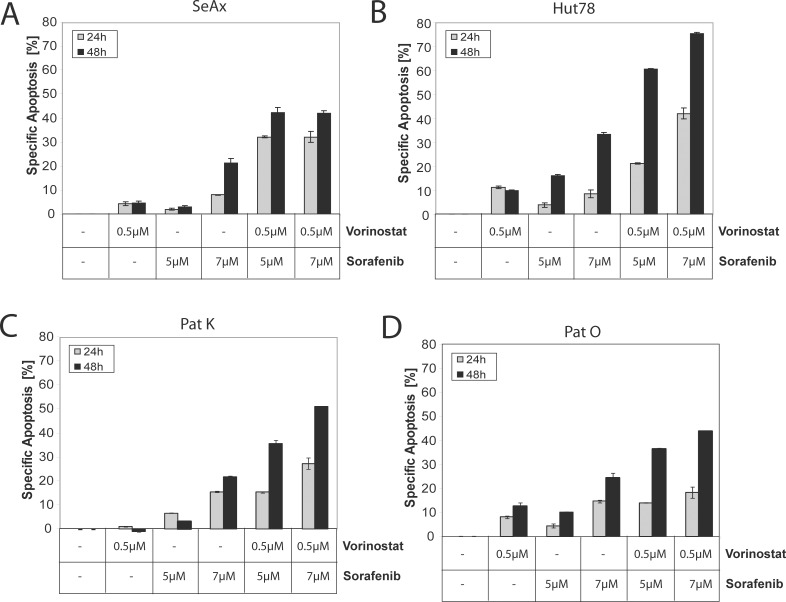
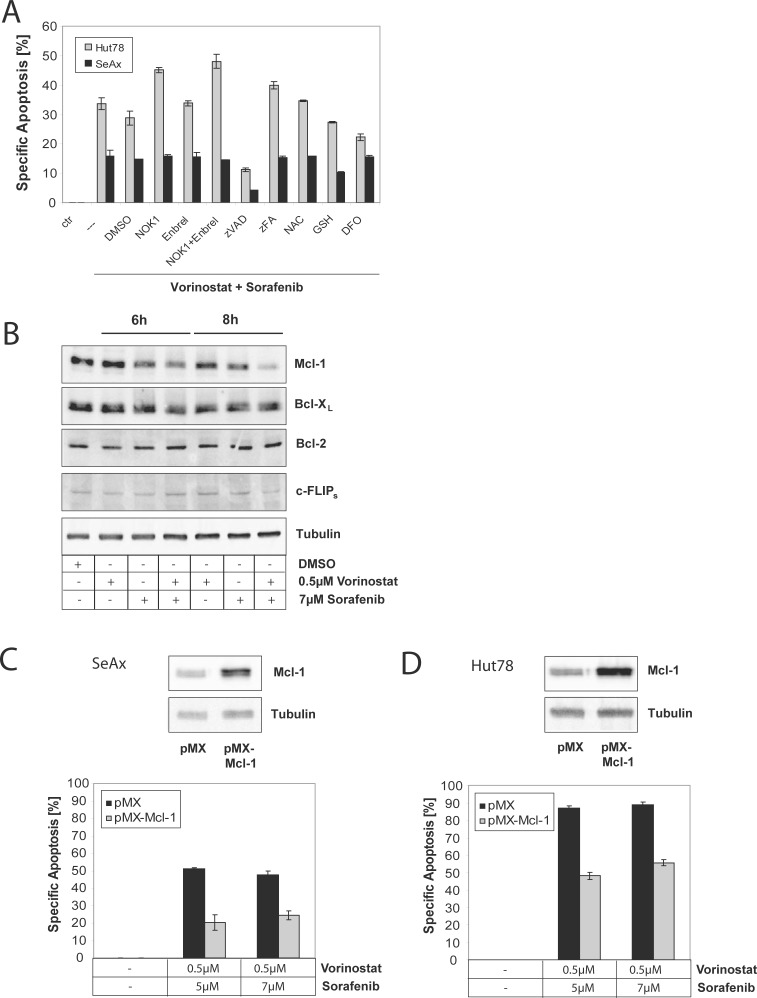
Therapy of cutaneous T cell lymphoma (CTCL) is complicated by a distinct resistance of the malignant T cells towards apoptosis that can be caused by NRAS mutations in late-stage patients. These mutations correlate with decreased overall survival, but sensitize the respective CTCL cells towards MEK-inhibition-induced apoptosis which represents a promising novel therapeutic target in CTCL. Here, we show that the multi-kinase inhibitor Sorafenib induces apoptosis in NRAS-mutated CTCL cells. CTCL cell lines and to a minor extent primary T cells from Sézary patients without NRAS mutations are also affected by Sorafenib-induced apoptosis suggesting a sensitizing role of NRAS mutations for Sorafenib-induced apoptosis. When combining Sorafenib with the established CTCL medication Vorinostat we detected an increase in cell death sensitivity in CTCL cells. The combination treatment acted synergistically in apoptosis induction in both non-mutant and mutant CTCL cells. Mechanistically, this synergistic apoptosis induction by Sorafenib and Vorinostat is based on the downregulation of the anti-apoptotic protein Mcl-1, but not of other Bcl-2 family members. Taken together, these findings suggest that Sorafenib in combination with Vorinostat represents a novel therapeutic approach for the treatment of CTCL patients.
Keywords: RAS mutation; T cell lymphoma; kinase; small molecule inhibitor; targeted therapy.
Conflict of interest statement
J.P.N. received travel and congress participation funding by TEVA as well as consulting fees by TEVA and Biogen. C.D.K. received travel support for scientific conferences and lecture fees for scientific presentations from TEVA/Cephalon Pharma GmbH and Therakos, Johnson & Johnson Medical GmbH. He was a member of the TEVA Cutaneous Lymphoma Advisory Board. P.H.K. and K.G. received consulting fees by the Biogen. The project was supported by the DKFZ / Bayer HealthCare Alliance.
Figures
References
-
- Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): part II. Prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223 e221–217. quiz 240–222. - PubMed
-
- Whittaker S, Hoppe R, Prince HM. How I treat mycosis fungoides and Sezary syndrome. Blood. 2016;127:3142–3153. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
