

An Introduction to Infinite HMMs for Single-Molecule Data Analysis
- PMID: 28538142
- PMCID: PMC5448313
- DOI: 10.1016/j.bpj.2017.04.027
An Introduction to Infinite HMMs for Single-Molecule Data Analysis
Abstract
The hidden Markov model (HMM) has been a workhorse of single-molecule data analysis and is now commonly used as a stand-alone tool in time series analysis or in conjunction with other analysis methods such as tracking. Here, we provide a conceptual introduction to an important generalization of the HMM, which is poised to have a deep impact across the field of biophysics: the infinite HMM (iHMM). As a modeling tool, iHMMs can analyze sequential data without a priori setting a specific number of states as required for the traditional (finite) HMM. Although the current literature on the iHMM is primarily intended for audiences in statistics, the idea is powerful and the iHMM's breadth in applicability outside machine learning and data science warrants a careful exposition. Here, we explain the key ideas underlying the iHMM, with a special emphasis on implementation, and provide a description of a code we are making freely available. In a companion article, we provide an important extension of the iHMM to accommodate complications such as drift.
Copyright © 2017 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures



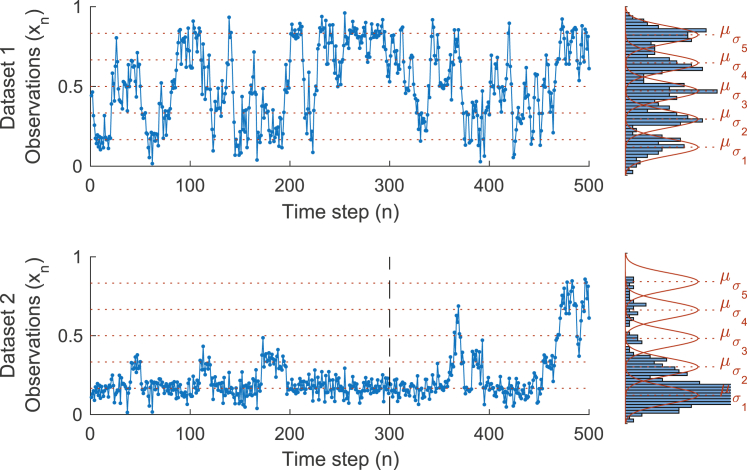


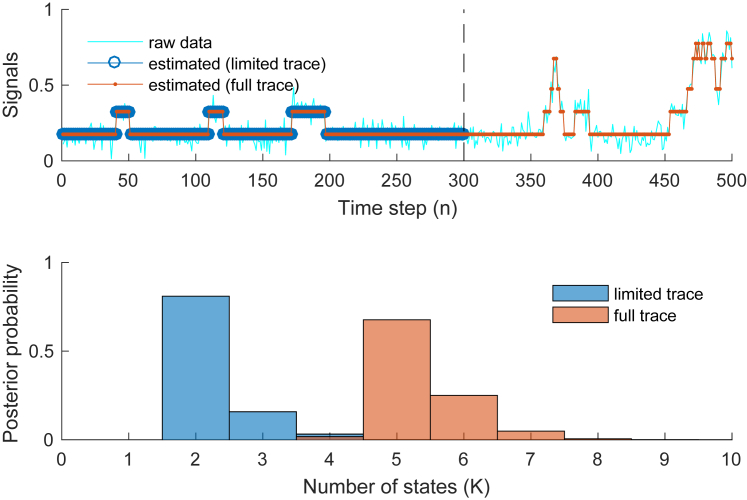
Similar articles
-
ICON: An Adaptation of Infinite HMMs for Time Traces with Drift.Biophys J. 2017 May 23;112(10):2117-2126. doi: 10.1016/j.bpj.2017.04.009. Biophys J. 2017. PMID: 28538149 Free PMC article.
-
Infinite hidden Markov models for unusual-event detection in video.IEEE Trans Image Process. 2008 May;17(5):811-22. doi: 10.1109/TIP.2008.919359. IEEE Trans Image Process. 2008. PMID: 18390385
-
Sparsely correlated hidden Markov models with application to genome-wide location studies.Bioinformatics. 2013 Mar 1;29(5):533-41. doi: 10.1093/bioinformatics/btt012. Epub 2013 Jan 16. Bioinformatics. 2013. PMID: 23325620 Free PMC article.
-
Hidden Markov model and its applications in motif findings.Methods Mol Biol. 2010;620:405-16. doi: 10.1007/978-1-60761-580-4_13. Methods Mol Biol. 2010. PMID: 20652513 Review.
-
Gaussian processes for machine learning.Int J Neural Syst. 2004 Apr;14(2):69-106. doi: 10.1142/S0129065704001899. Int J Neural Syst. 2004. PMID: 15112367 Review.
Cited by
-
Direct observation of Thermomyces lanuginosus lipase diffusional states by Single Particle Tracking and their remodeling by mutations and inhibition.Sci Rep. 2019 Nov 7;9(1):16169. doi: 10.1038/s41598-019-52539-1. Sci Rep. 2019. PMID: 31700110 Free PMC article.
-
Phosphorylation Induces Conformational Rigidity at the C-Terminal Domain of AMPA Receptors.J Phys Chem B. 2019 Jan 10;123(1):130-137. doi: 10.1021/acs.jpcb.8b10749. Epub 2018 Dec 27. J Phys Chem B. 2019. PMID: 30537817 Free PMC article.
-
Inferring effective forces for Langevin dynamics using Gaussian processes.J Chem Phys. 2020 Mar 31;152(12):124106. doi: 10.1063/1.5144523. J Chem Phys. 2020. PMID: 32241120 Free PMC article.
-
AutoStepfinder: A fast and automated step detection method for single-molecule analysis.Patterns (N Y). 2021 Apr 30;2(5):100256. doi: 10.1016/j.patter.2021.100256. eCollection 2021 May 14. Patterns (N Y). 2021. PMID: 34036291 Free PMC article.
-
Bayesian inference of kinetic schemes for ion channels by Kalman filtering.Elife. 2022 May 4;11:e62714. doi: 10.7554/eLife.62714. Elife. 2022. PMID: 35506659 Free PMC article.
References
-
- Rabiner L., Juang B. An introduction to hidden Markov models. IEEE ASSP Mag. 1986;3:4–16.
-
- Eddy S.R. What is a hidden Markov model? Nat. Biotechnol. 2004;22:1315–1316. - PubMed
-
- Krogh A., Brown M., Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J. Mol. Biol. 1994;235:1501–1531. - PubMed
-
- Streit R.L., Barrett R.F. Frequency line tracking using hidden Markov models. IEEE Trans. Acoust. Speech Signal Process. 1990;38:586–598.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
