

Taxane-Platin-Resistant Lung Cancers Co-develop Hypersensitivity to JumonjiC Demethylase Inhibitors
- PMID: 28538184
- PMCID: PMC5531293
- DOI: 10.1016/j.celrep.2017.04.077
Taxane-Platin-Resistant Lung Cancers Co-develop Hypersensitivity to JumonjiC Demethylase Inhibitors
Abstract
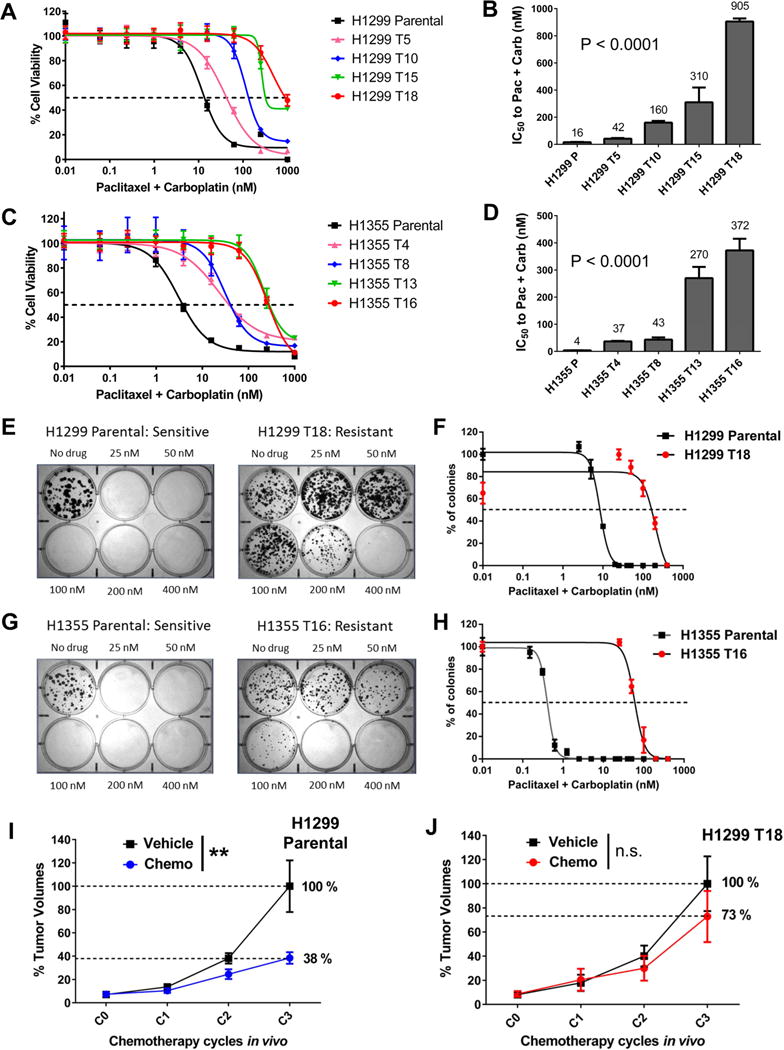
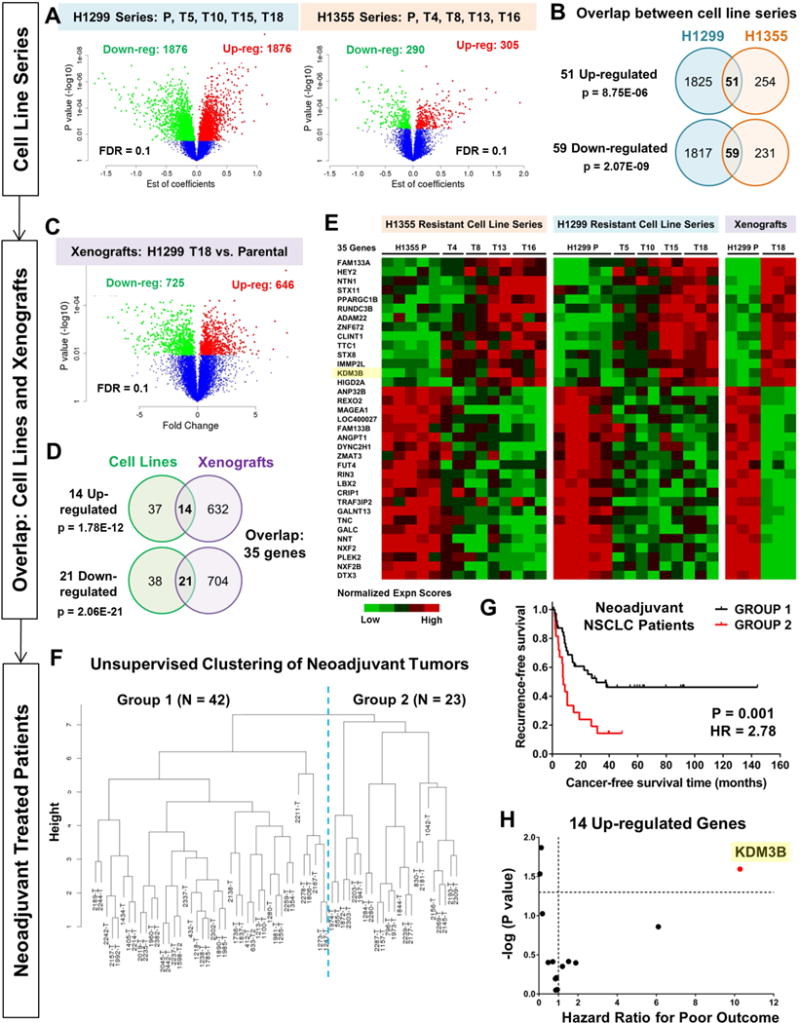
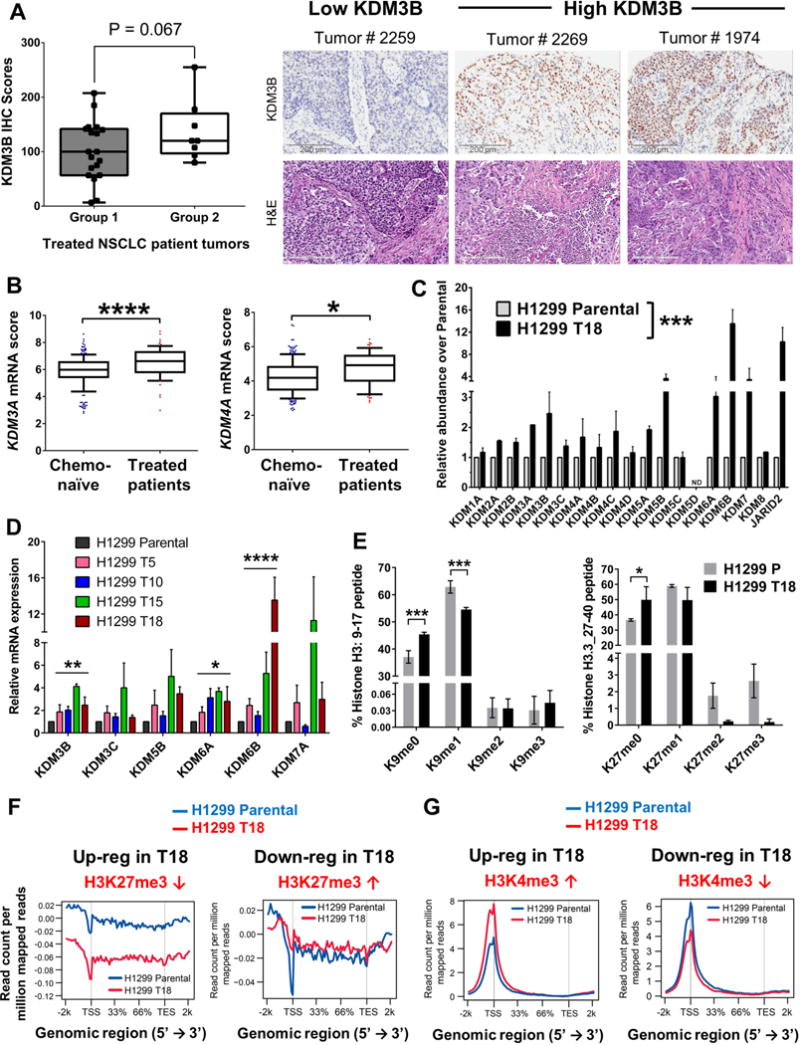
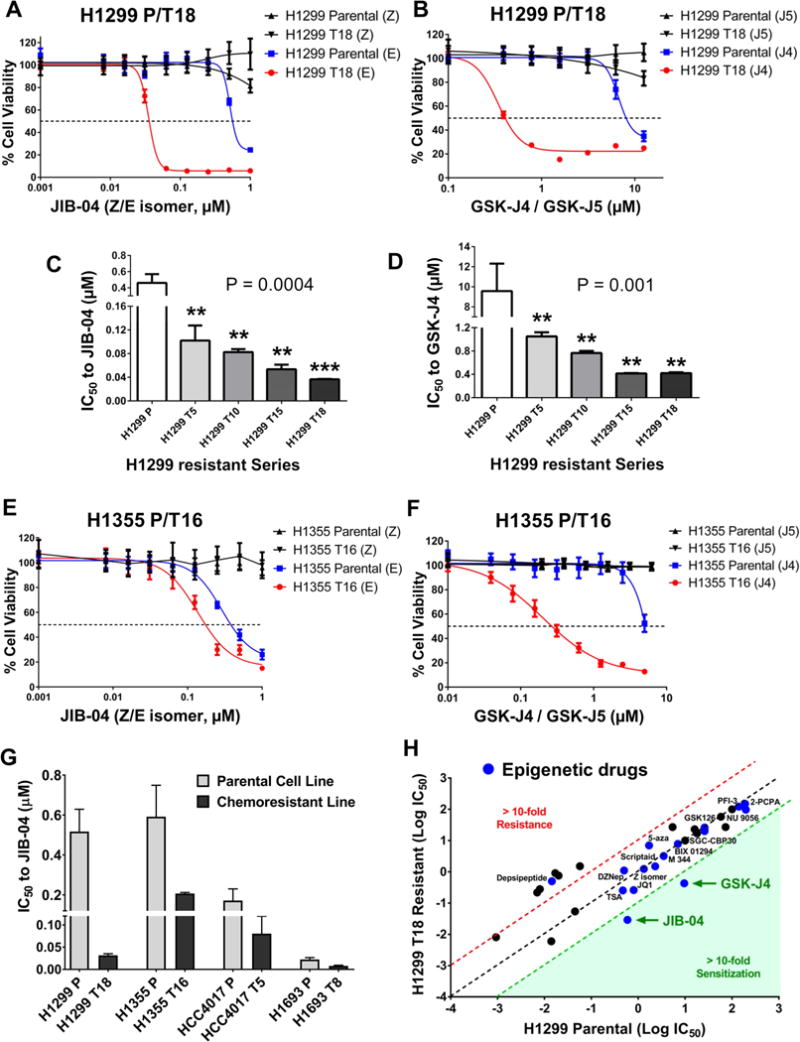
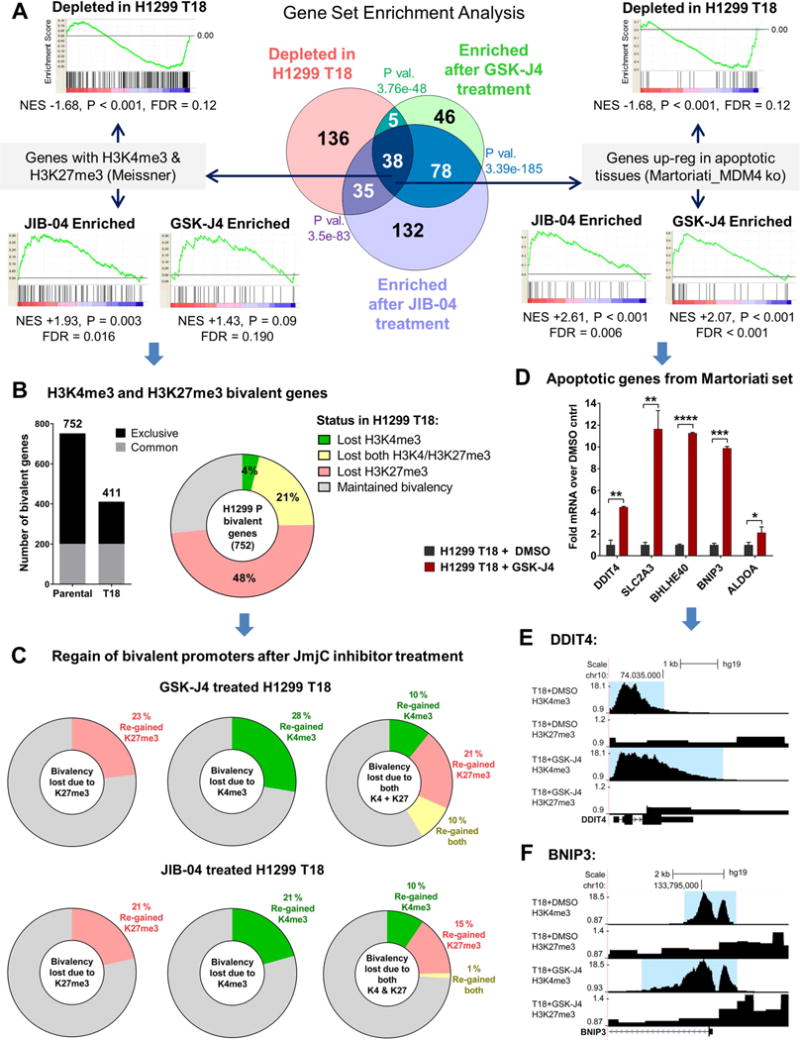
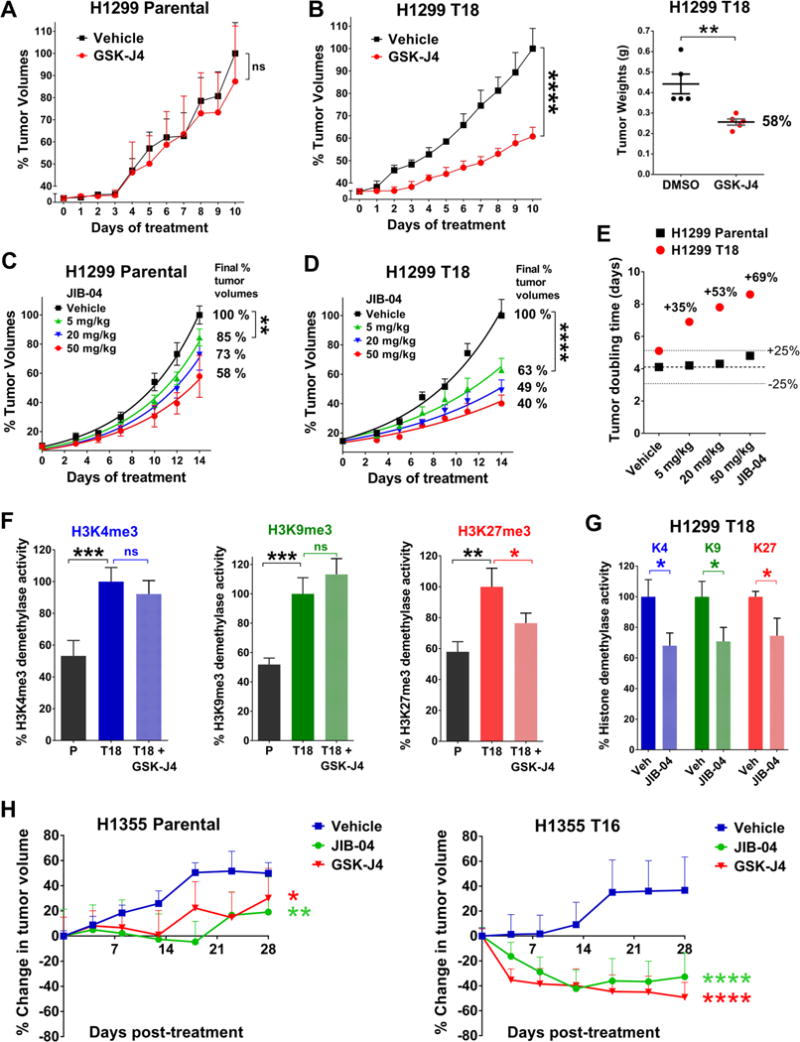
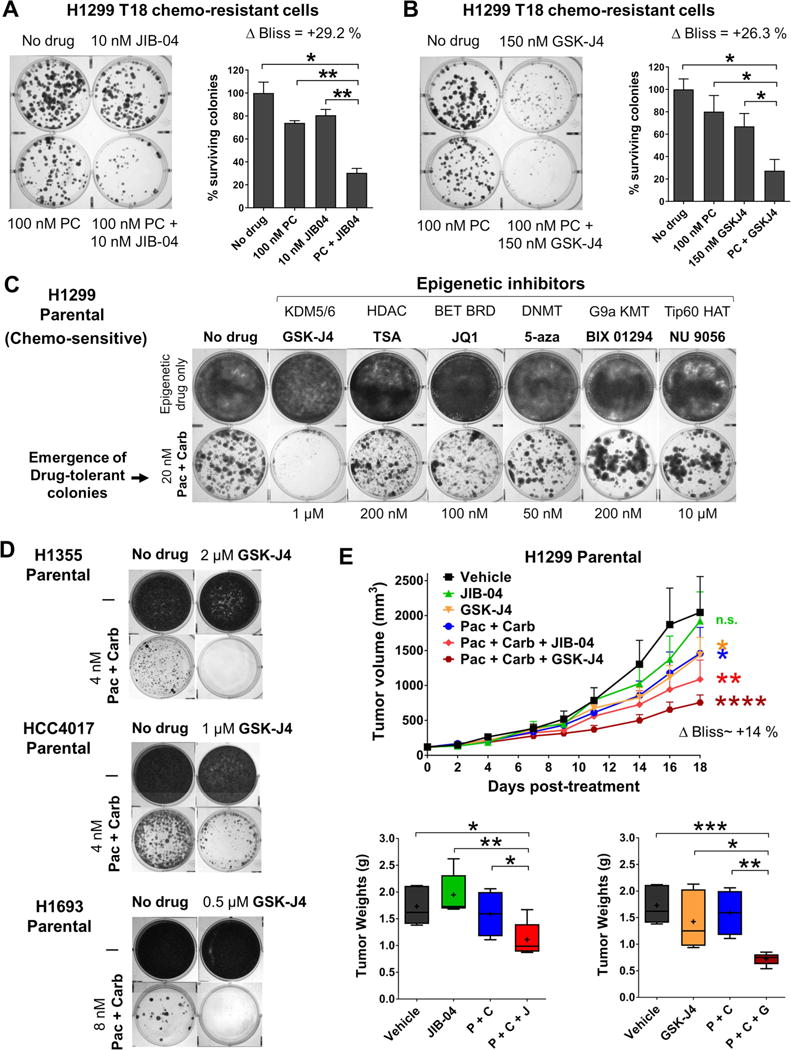
Although non-small cell lung cancer (NSCLC) patients benefit from standard taxane-platin chemotherapy, many relapse, developing drug resistance. We established preclinical taxane-platin-chemoresistance models and identified a 35-gene resistance signature, which was associated with poor recurrence-free survival in neoadjuvant-treated NSCLC patients and included upregulation of the JumonjiC lysine demethylase KDM3B. In fact, multi-drug-resistant cells progressively increased the expression of many JumonjiC demethylases, had altered histone methylation, and, importantly, showed hypersensitivity to JumonjiC inhibitors in vitro and in vivo. Increasing taxane-platin resistance in progressive cell line series was accompanied by progressive sensitization to JIB-04 and GSK-J4. These JumonjiC inhibitors partly reversed deregulated transcriptional programs, prevented the emergence of drug-tolerant colonies from chemo-naive cells, and synergized with standard chemotherapy in vitro and in vivo. Our findings reveal JumonjiC inhibitors as promising therapies for targeting taxane-platin-chemoresistant NSCLCs.
Keywords: GSK-J4; JIB-04; Jumonji demethylases; KDM; demethylase inhibitors; drug resistance; histone demethylases; histone methylation; lung cancer; taxane-platin chemotherapy.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- American Cancer Society. Cancer Facts & Figures. 2015.
-
- Bradshaw DM, Arceci RJ. Clinical relevance of transmembrane drug efflux as a mechanism of multidrug resistance. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1998;16:3674–3690. - PubMed
-
- Busschots S, O’Toole S, O’Leary JJ, Stordal B. Carboplatin and taxol resistance develops more rapidly in functional BRCA1 compared to dysfunctional BRCA1 ovarian cancer cells. Experimental cell research. 2015;336:1–14. - PubMed
-
- d’Amato TA, Landreneau RJ, Ricketts W, Huang W, Parker R, Mechetner E, Yu IR, Luketich JD. Chemotherapy resistance and oncogene expression in non-small cell lung cancer. The Journal of thoracic and cardiovascular surgery. 2007;133:352–363. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
