

Applying systems-level spectral imaging and analysis to reveal the organelle interactome
- PMID: 28538724
- PMCID: PMC5536967
- DOI: 10.1038/nature22369
Applying systems-level spectral imaging and analysis to reveal the organelle interactome
Abstract
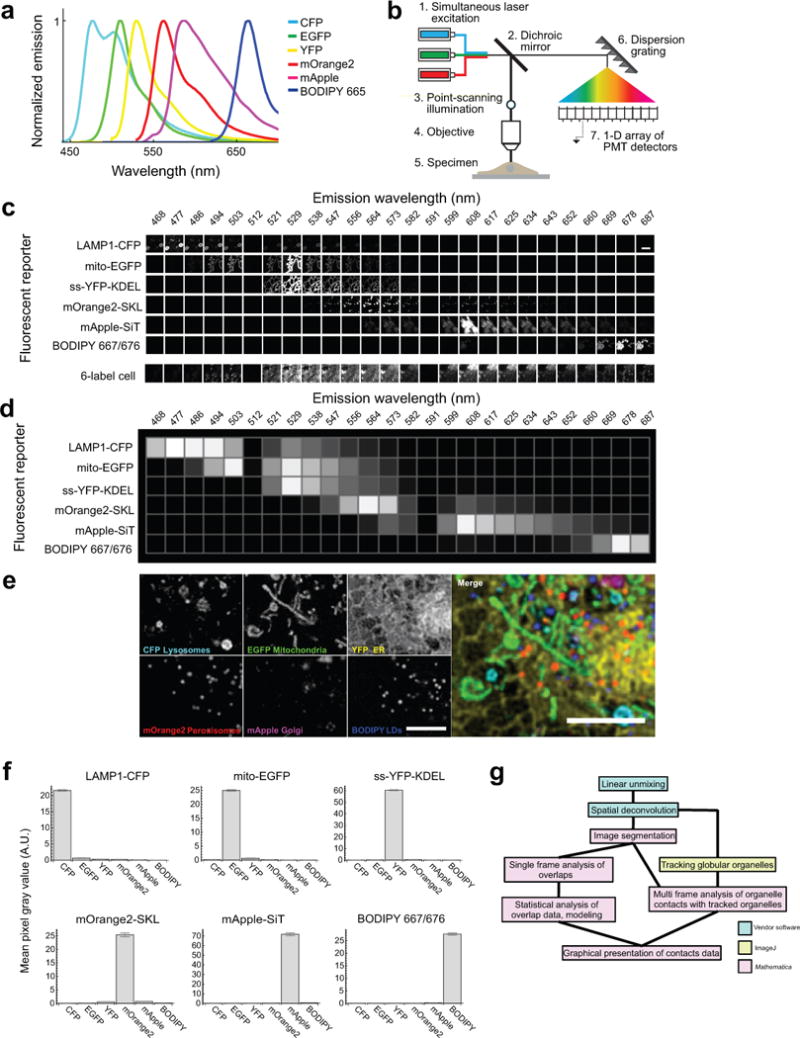
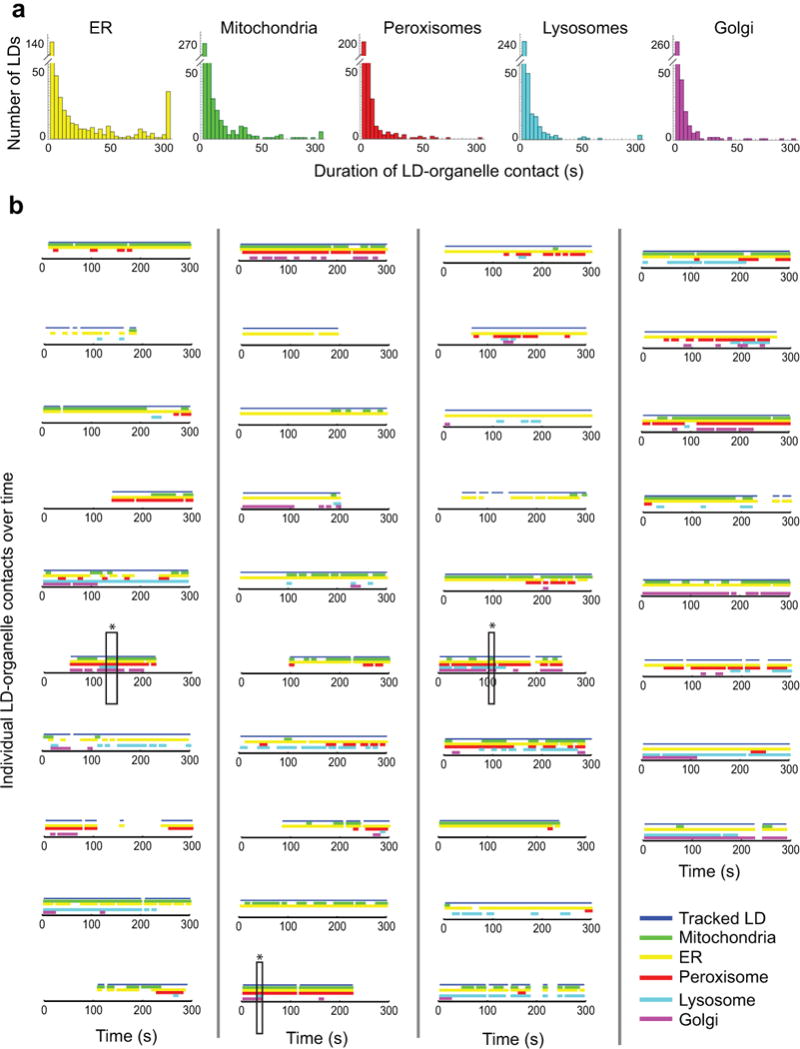
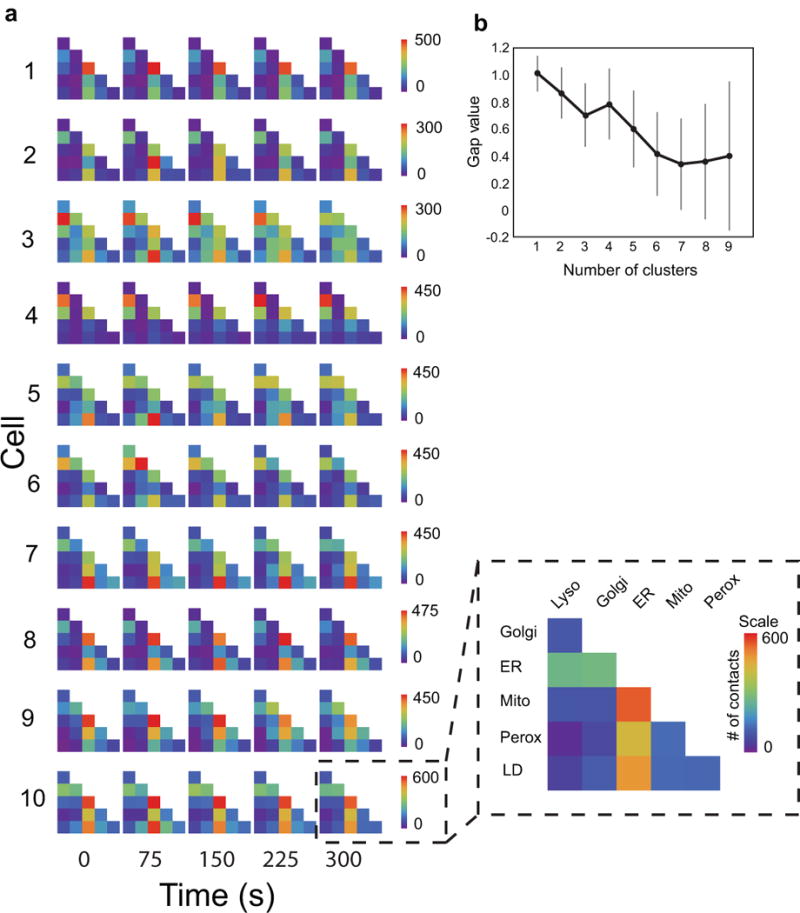
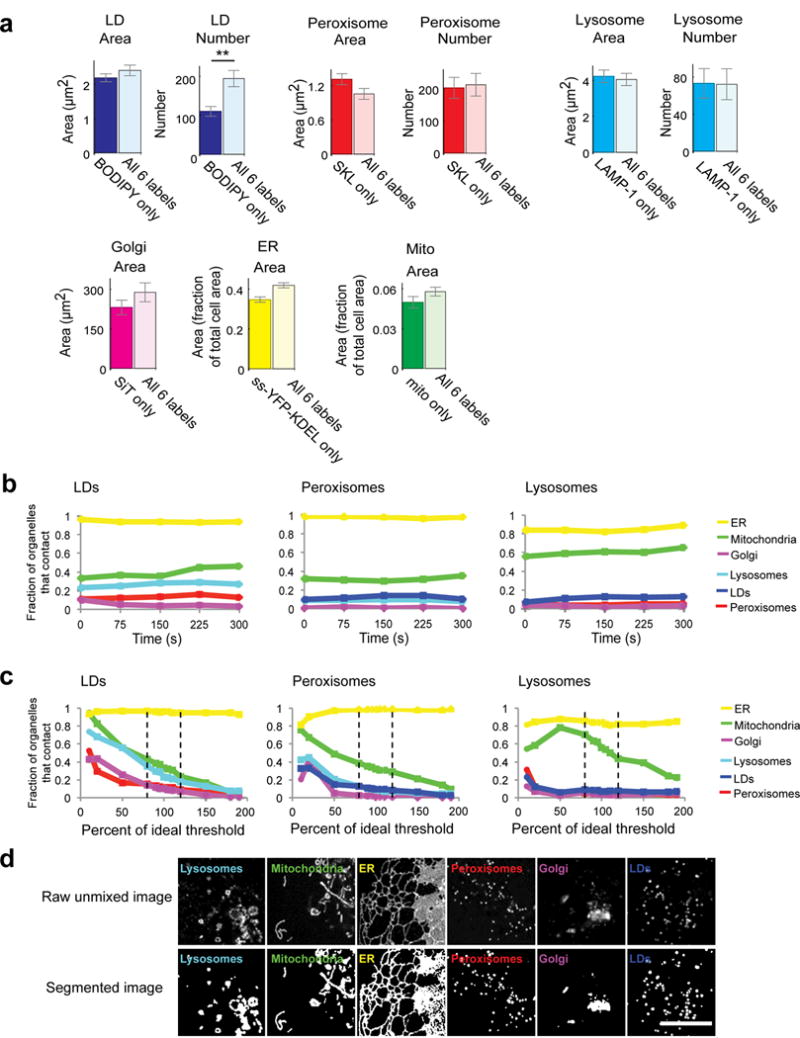
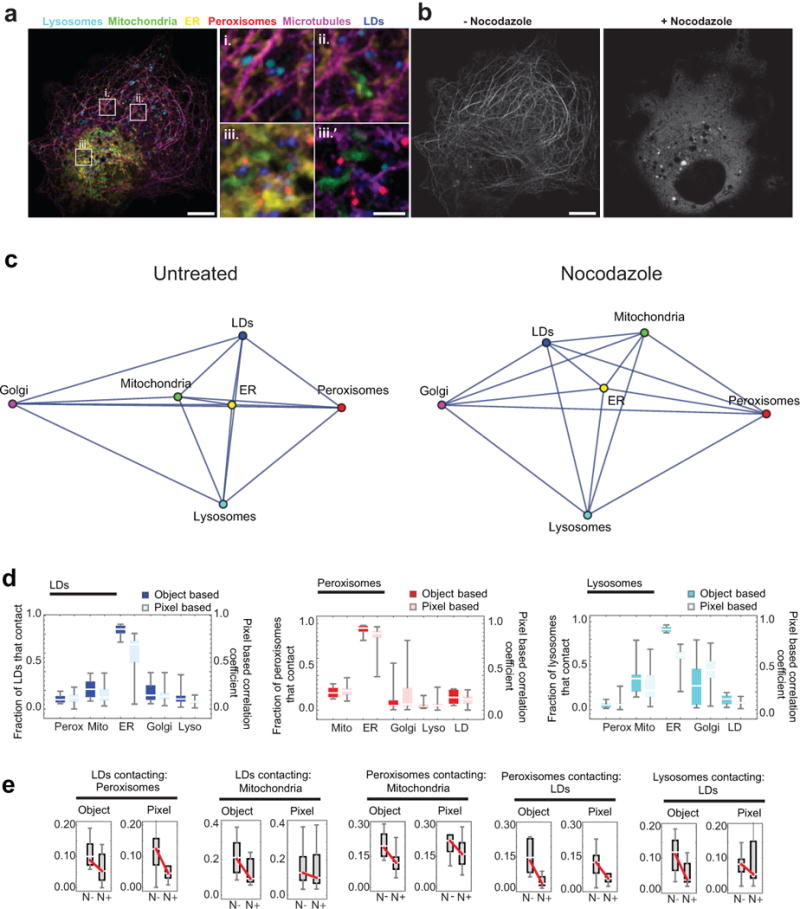
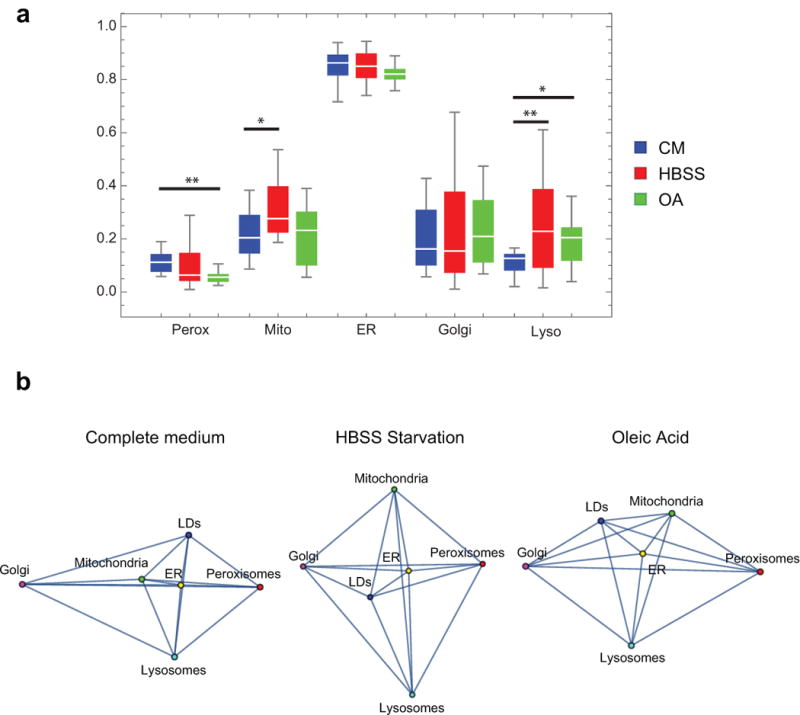
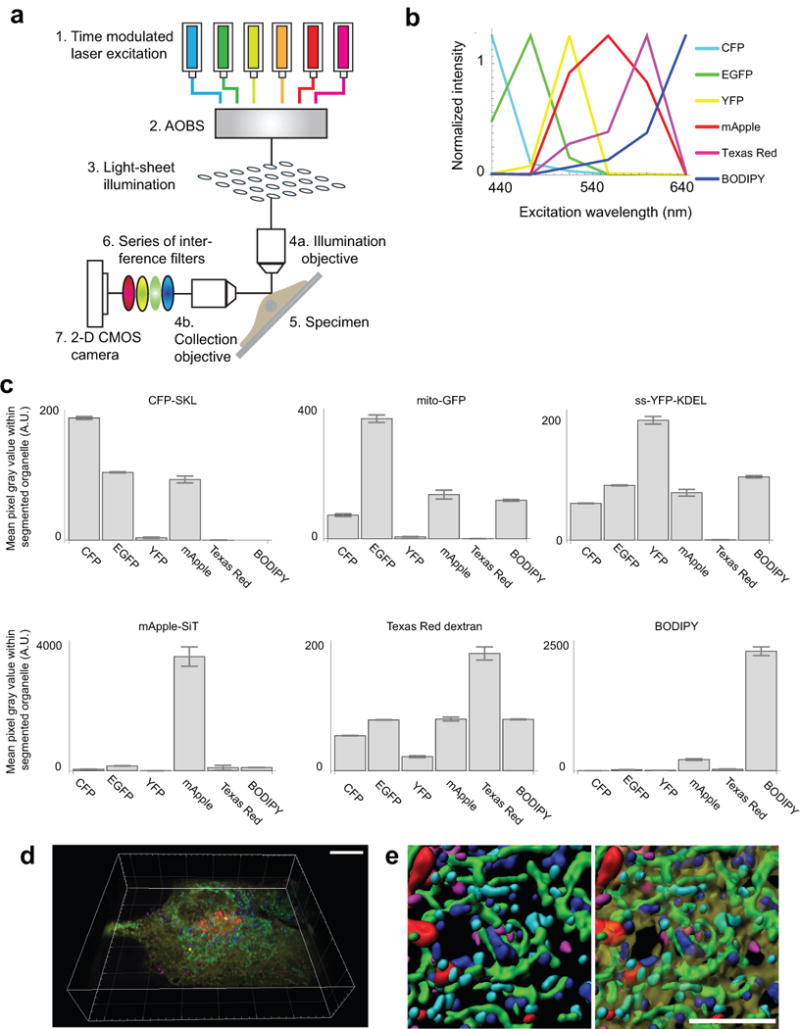
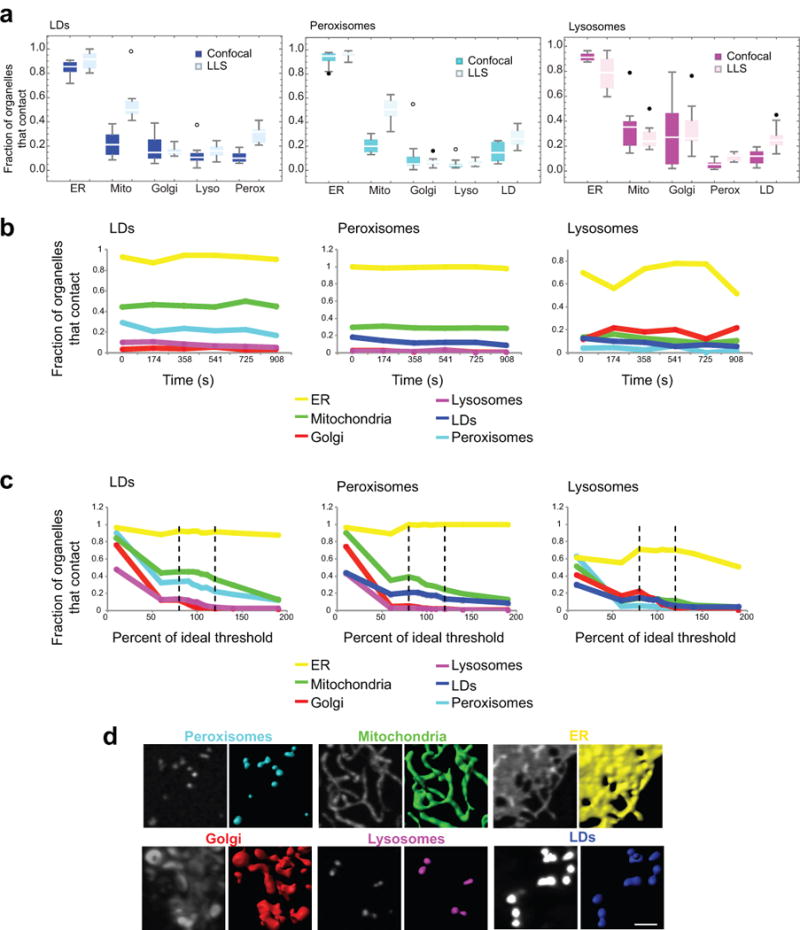
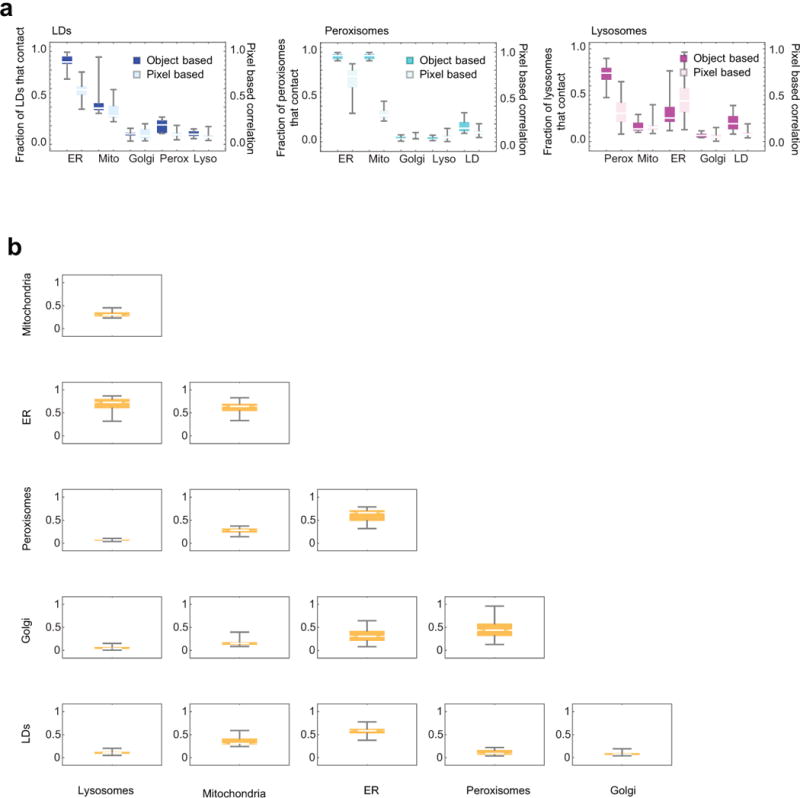
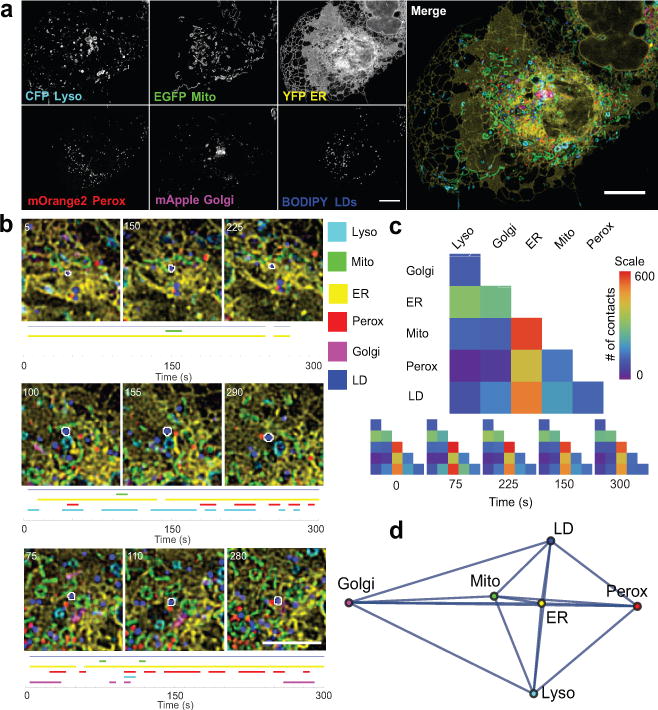
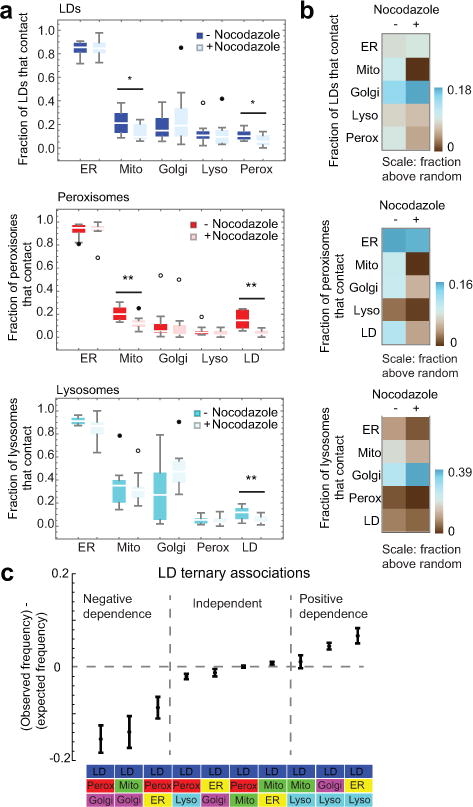
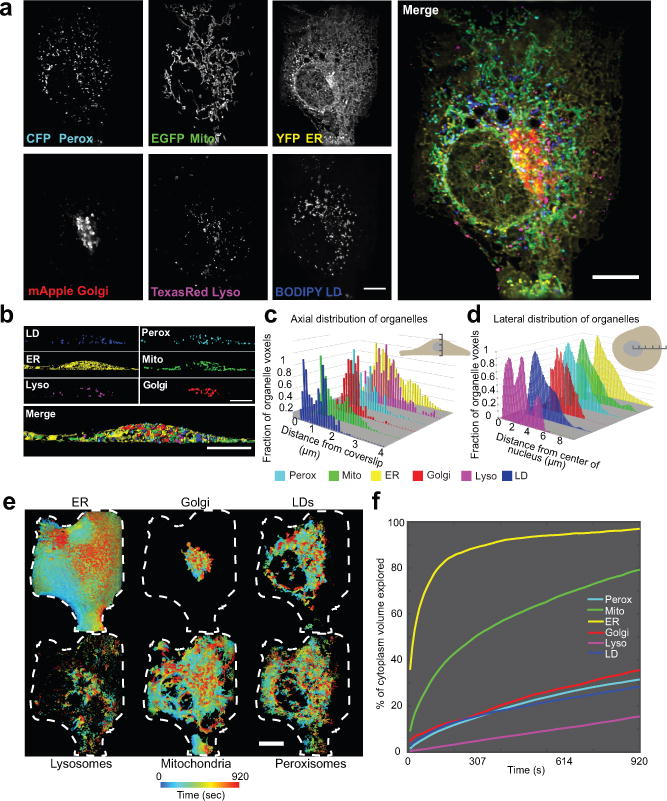
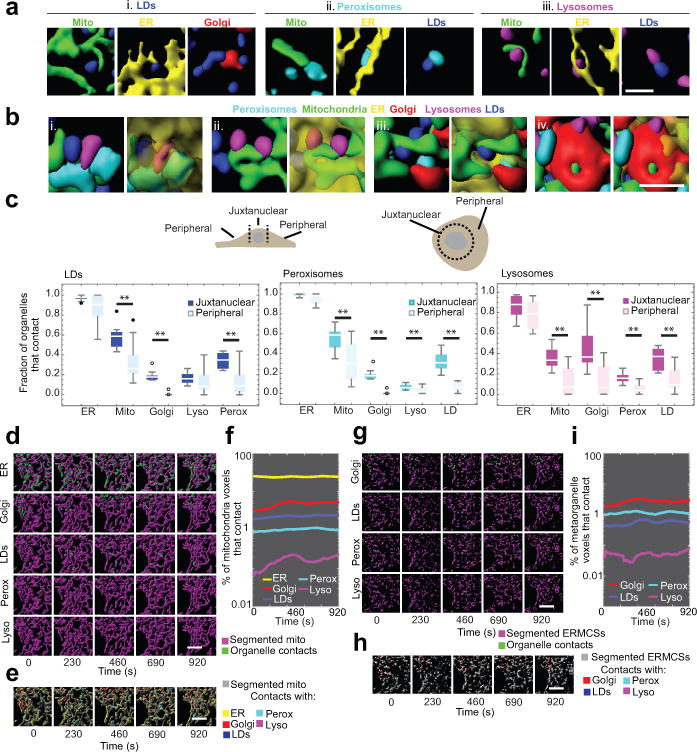
The organization of the eukaryotic cell into discrete membrane-bound organelles allows for the separation of incompatible biochemical processes, but the activities of these organelles must be coordinated. For example, lipid metabolism is distributed between the endoplasmic reticulum for lipid synthesis, lipid droplets for storage and transport, mitochondria and peroxisomes for β-oxidation, and lysosomes for lipid hydrolysis and recycling. It is increasingly recognized that organelle contacts have a vital role in diverse cellular functions. However, the spatial and temporal organization of organelles within the cell remains poorly characterized, as fluorescence imaging approaches are limited in the number of different labels that can be distinguished in a single image. Here we present a systems-level analysis of the organelle interactome using a multispectral image acquisition method that overcomes the challenge of spectral overlap in the fluorescent protein palette. We used confocal and lattice light sheet instrumentation and an imaging informatics pipeline of five steps to achieve mapping of organelle numbers, volumes, speeds, positions and dynamic inter-organelle contacts in live cells from a monkey fibroblast cell line. We describe the frequency and locality of two-, three-, four- and five-way interactions among six different membrane-bound organelles (endoplasmic reticulum, Golgi, lysosome, peroxisome, mitochondria and lipid droplet) and show how these relationships change over time. We demonstrate that each organelle has a characteristic distribution and dispersion pattern in three-dimensional space and that there is a reproducible pattern of contacts among the six organelles, that is affected by microtubule and cell nutrient status. These live-cell confocal and lattice light sheet spectral imaging approaches are applicable to any cell system expressing multiple fluorescent probes, whether in normal conditions or when cells are exposed to disturbances such as drugs, pathogens or stress. This methodology thus offers a powerful descriptive tool and can be used to develop hypotheses about cellular organization and dynamics.
Conflict of interest statement
Figures
Comment in
-
Cell imaging: An intracellular dance visualized.Nature. 2017 Jun 1;546(7656):39-40. doi: 10.1038/nature22500. Epub 2017 May 24. Nature. 2017. PMID: 28538721 No abstract available.
References
Extended Data References
-
- Garini Y, Young IT, McNamara G. Spectral imaging: Principles and applications. Cytometry A. 2006;69A:735–747. - PubMed
-
- Tsuriel S, Gudes S, Draft RW, Binshtok AM, Lichtman JW. Multispectral labeling technique to map many neighboring axonal projections in the same tissue. Nat Methods. 2015;12:547–552. - PubMed
-
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. - PubMed
