

Comprehensive whole genome sequence analyses yields novel genetic and structural insights for Intellectual Disability
- PMID: 28539120
- PMCID: PMC5442678
- DOI: 10.1186/s12864-017-3671-0
Comprehensive whole genome sequence analyses yields novel genetic and structural insights for Intellectual Disability
Abstract
Background: Intellectual Disability (ID) is among the most common global disorders, yet etiology is unknown in ~30% of patients despite clinical assessment. Whole genome sequencing (WGS) is able to interrogate the entire genome, providing potential to diagnose idiopathic patients.
Methods: We conducted WGS on eight children with idiopathic ID and brain structural defects, and their normal parents; carrying out an extensive data analyses, using standard and discovery approaches.
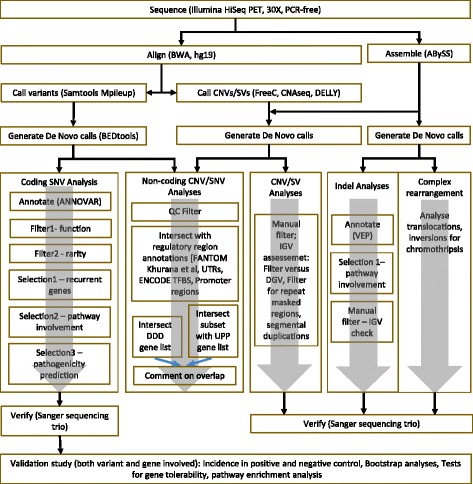
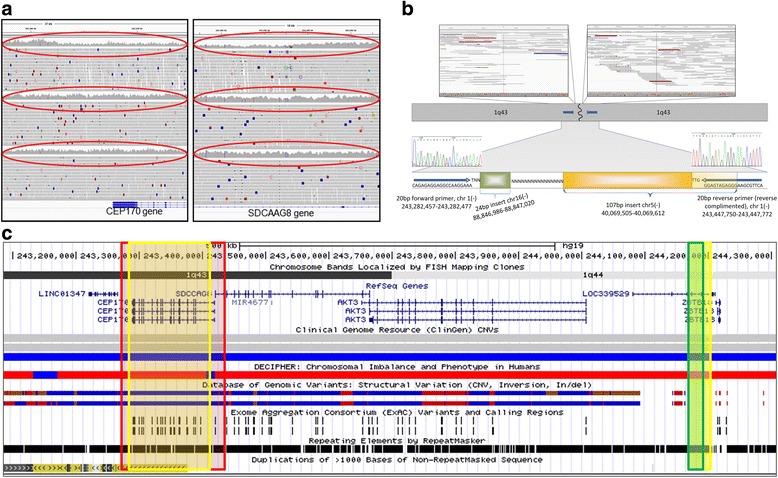
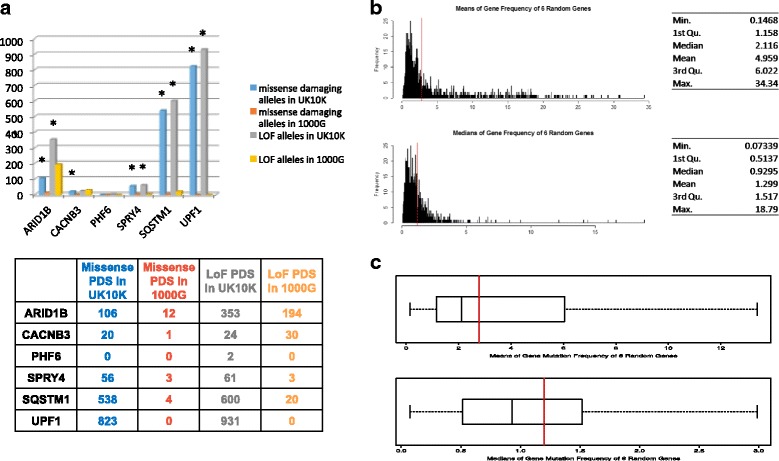
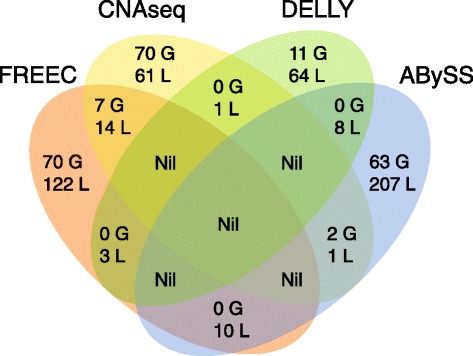
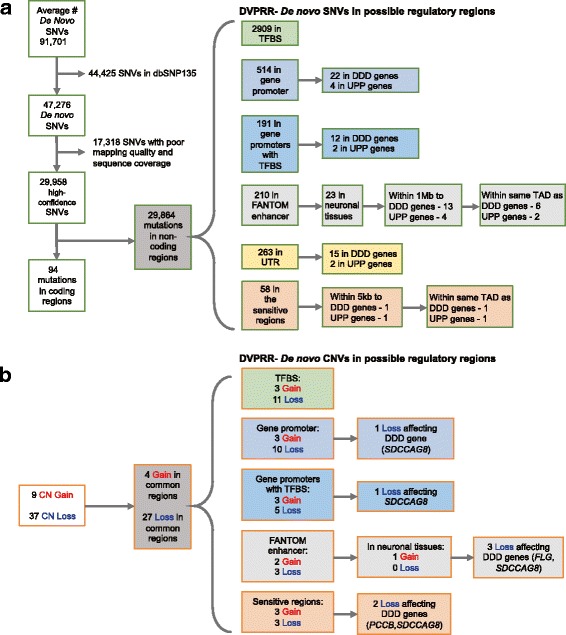
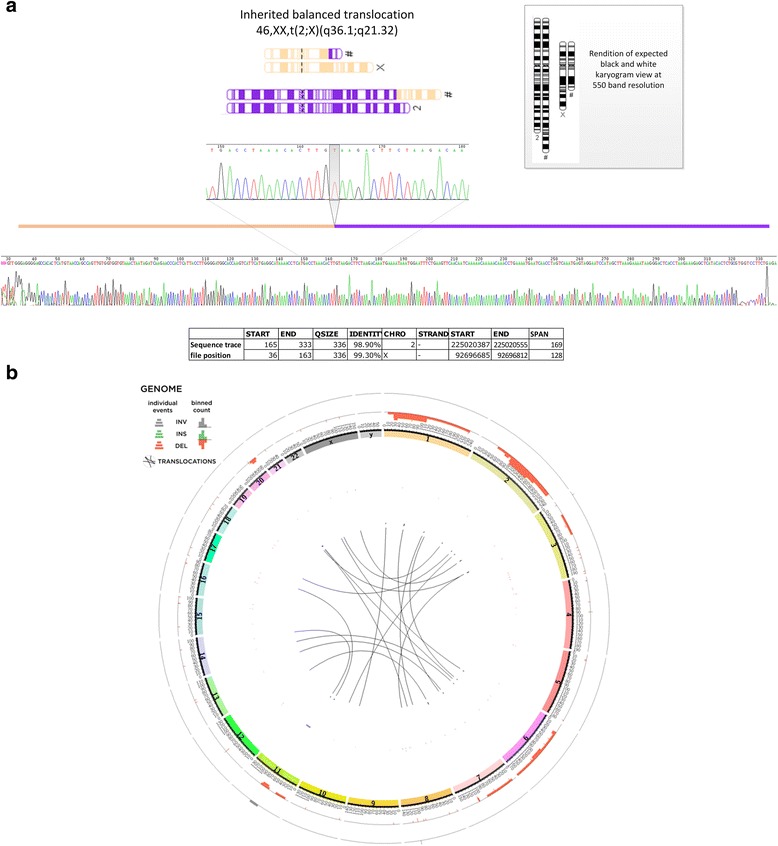
Results: We verified de novo pathogenic single nucleotide variants (SNV) in ARID1B c.1595delG and PHF6 c.820C > T, potentially causative de novo two base indels in SQSTM1 c.115_116delinsTA and UPF1 c.1576_1577delinsA, and de novo SNVs in CACNB3 c.1289G > A, and SPRY4 c.508 T > A, of uncertain significance. We report results from a large secondary control study of 2081 exomes probing the pathogenicity of the above genes. We analyzed structural variation by four different algorithms including de novo genome assembly. We confirmed a likely contributory 165 kb de novo heterozygous 1q43 microdeletion missed by clinical microarray. The de novo assembly resulted in unmasking hidden genome instability that was missed by standard re-alignment based algorithms. We also interrogated regulatory sequence variation for known and hypothesized ID genes and present useful strategies for WGS data analyses for non-coding variation.
Conclusion: This study provides an extensive analysis of WGS in the context of ID, providing genetic and structural insights into ID and yielding diagnoses.
Keywords: 1q43 microdeletion; ARID1B; CACNB3; Genome assembly; Intellectual Disability; PHF6; SPRY4; SQSTM1; UPF1; Whole genome sequencing.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
