

HIV-1 full-genome phylogenetics of generalized epidemics in sub-Saharan Africa: impact of missing nucleotide characters in next-generation sequences
- PMID: 28540766
- PMCID: PMC5597042
- DOI: 10.1089/AID.2017.0061
HIV-1 full-genome phylogenetics of generalized epidemics in sub-Saharan Africa: impact of missing nucleotide characters in next-generation sequences
Abstract
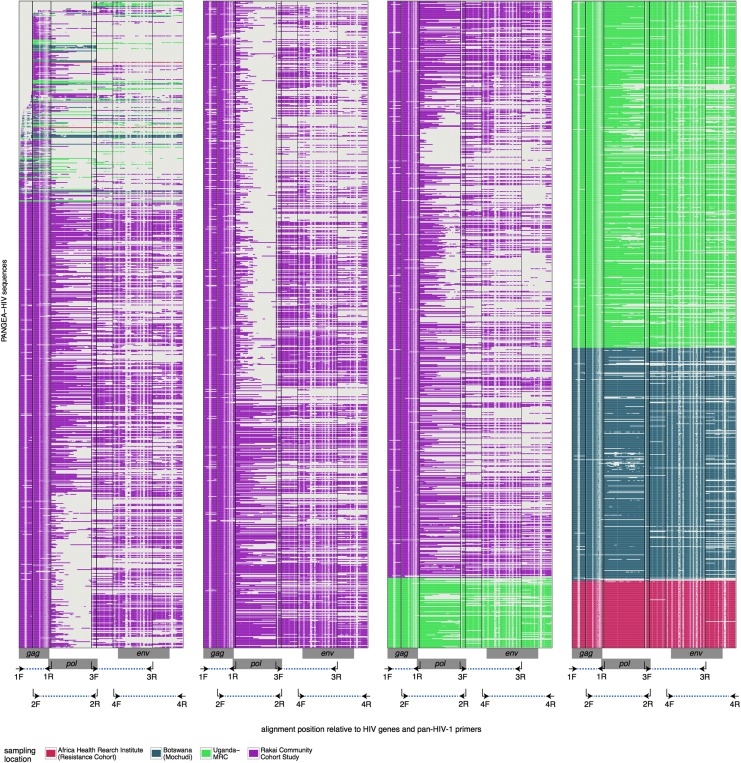
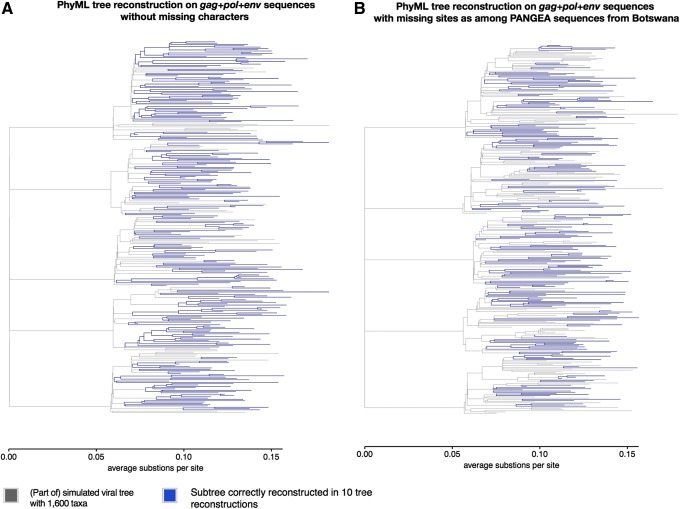
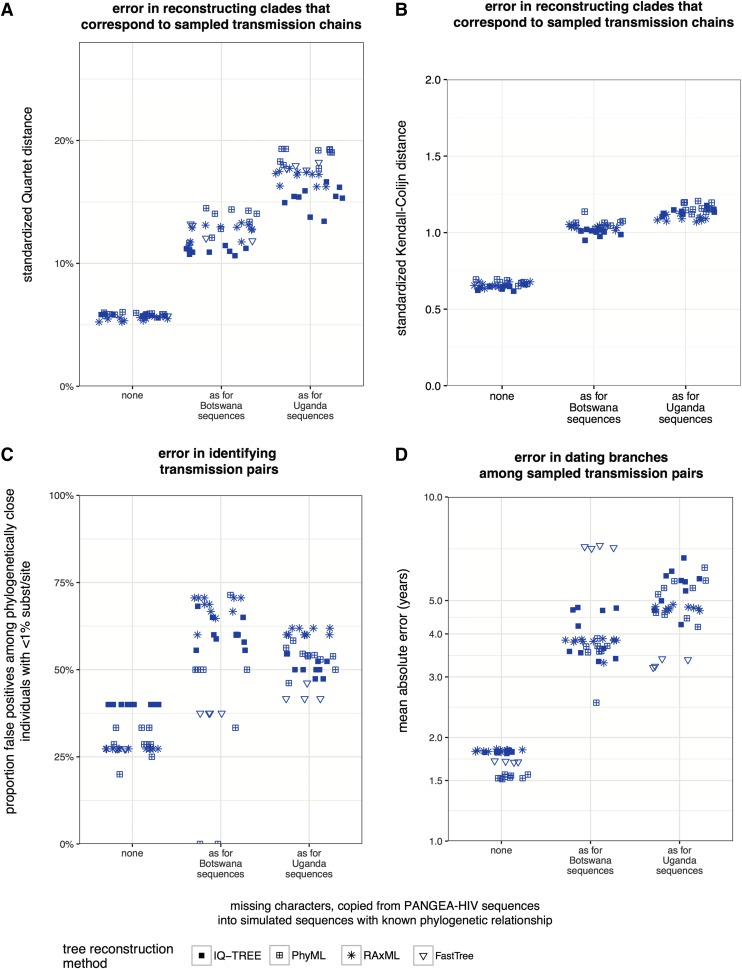
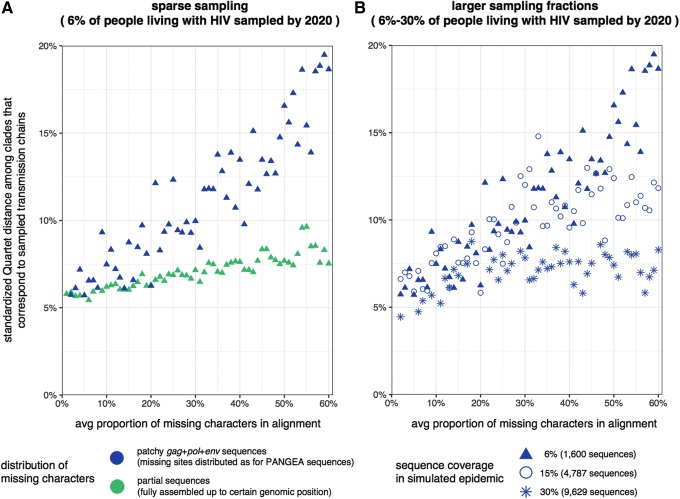
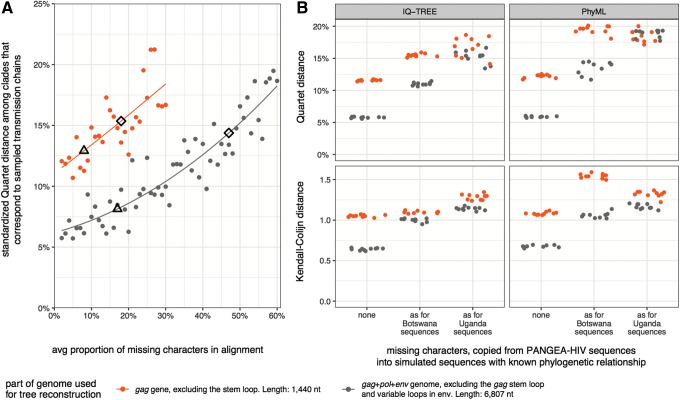
To characterize HIV-1 transmission dynamics in regions where the burden of HIV-1 is greatest, the 'Phylogenetics and Networks for Generalised HIV Epidemics in Africa' consortium (PANGEA-HIV) is sequencing full-genome viral isolates from across sub-Saharan Africa. We report the first 3,985 PANGEA-HIV consensus sequences from four cohort sites (Rakai Community Cohort Study, n=2,833; MRC/UVRI Uganda, n=701; Mochudi Prevention Project, n=359; Africa Health Research Institute Resistance Cohort, n=92). Next-generation sequencing success rates varied: more than 80% of the viral genome from the gag to the nef genes could be determined for all sequences from South Africa, 75% of sequences from Mochudi, 60% of sequences from MRC/UVRI Uganda, and 22% of sequences from Rakai. Partial sequencing failure was primarily associated with low viral load, increased for amplicons closer to the 3' end of the genome, was not associated with subtype diversity except HIV-1 subtype D, and remained significantly associated with sampling location after controlling for other factors. We assessed the impact of the missing data patterns in PANGEA-HIV sequences on phylogeny reconstruction in simulations. We found a threshold in terms of taxon sampling below which the patchy distribution of missing characters in next-generation sequences has an excess negative impact on the accuracy of HIV-1 phylogeny reconstruction, which is attributable to tree reconstruction artifacts that accumulate when branches in viral trees are long. The large number of PANGEA-HIV sequences provides unprecedented opportunities for evaluating HIV-1 transmission dynamics across sub-Saharan Africa and identifying prevention opportunities. Molecular epidemiological analyses of these data must proceed cautiously because sequence sampling remains below the identified threshold and a considerable negative impact of missing characters on phylogeny reconstruction is expected.
Conflict of interest statement
No competing financial interests exist.
Figures
Similar articles
-
PANGEA-HIV 2: Phylogenetics And Networks for Generalised Epidemics in Africa.Curr Opin HIV AIDS. 2019 May;14(3):173-180. doi: 10.1097/COH.0000000000000542. Curr Opin HIV AIDS. 2019. PMID: 30946141 Free PMC article. Review.
-
Phylogenetic Tools for Generalized HIV-1 Epidemics: Findings from the PANGEA-HIV Methods Comparison.Mol Biol Evol. 2017 Jan;34(1):185-203. doi: 10.1093/molbev/msw217. Epub 2016 Oct 7. Mol Biol Evol. 2017. PMID: 28053012 Free PMC article.
-
Recombination Analysis of Near Full-Length HIV-1 Sequences and the Identification of a Potential New Circulating Recombinant Form from Rakai, Uganda.AIDS Res Hum Retroviruses. 2020 Jun;36(6):467-474. doi: 10.1089/AID.2019.0150. Epub 2020 Mar 2. AIDS Res Hum Retroviruses. 2020. PMID: 31914792 Free PMC article.
-
Using nearly full-genome HIV sequence data improves phylogeny reconstruction in a simulated epidemic.Sci Rep. 2016 Dec 23;6:39489. doi: 10.1038/srep39489. Sci Rep. 2016. PMID: 28008945 Free PMC article.
-
Phylogenetic studies of transmission dynamics in generalized HIV epidemics: an essential tool where the burden is greatest?J Acquir Immune Defic Syndr. 2014 Oct 1;67(2):181-95. doi: 10.1097/QAI.0000000000000271. J Acquir Immune Defic Syndr. 2014. PMID: 24977473 Free PMC article. Review.
Cited by
-
Genetic Cluster Analysis for HIV Prevention.Curr HIV/AIDS Rep. 2018 Apr;15(2):182-189. doi: 10.1007/s11904-018-0384-1. Curr HIV/AIDS Rep. 2018. PMID: 29460226 Free PMC article. Review.
-
Longitudinal population-level HIV epidemiologic and genomic surveillance highlights growing gender disparity of HIV transmission in Uganda.Nat Microbiol. 2024 Jan;9(1):35-54. doi: 10.1038/s41564-023-01530-8. Epub 2023 Dec 5. Nat Microbiol. 2024. PMID: 38052974 Free PMC article.
-
Effect of HIV Subtype and Antiretroviral Therapy on HIV-Associated Neurocognitive Disorder Stage in Rakai, Uganda.J Acquir Immune Defic Syndr. 2019 Jun 1;81(2):216-223. doi: 10.1097/QAI.0000000000001992. J Acquir Immune Defic Syndr. 2019. PMID: 30865184 Free PMC article.
-
Phylodynamic Structure in the Botswana HIV Epidemic.Res Sq [Preprint]. 2024 Oct 18:rs.3.rs-4969814. doi: 10.21203/rs.3.rs-4969814/v1. Res Sq. 2024. PMID: 39483888 Free PMC article. Preprint.
-
Prediction of Coreceptor Tropism in HIV-1 Subtype C in Botswana.Viruses. 2023 Jan 31;15(2):403. doi: 10.3390/v15020403. Viruses. 2023. PMID: 36851617 Free PMC article.
References
-
- Brenner BG, Roger M, Routy JP, Moisi D, Ntemgwa M, Matte C, et al. : High rates of forward transmission events after acute/early HIV-1 infection. J Infect Dis 2007;195:951–959 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources