

Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations
- PMID: 28541286
- PMCID: PMC5477518
- DOI: 10.1038/ncomms15277
Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations
Abstract
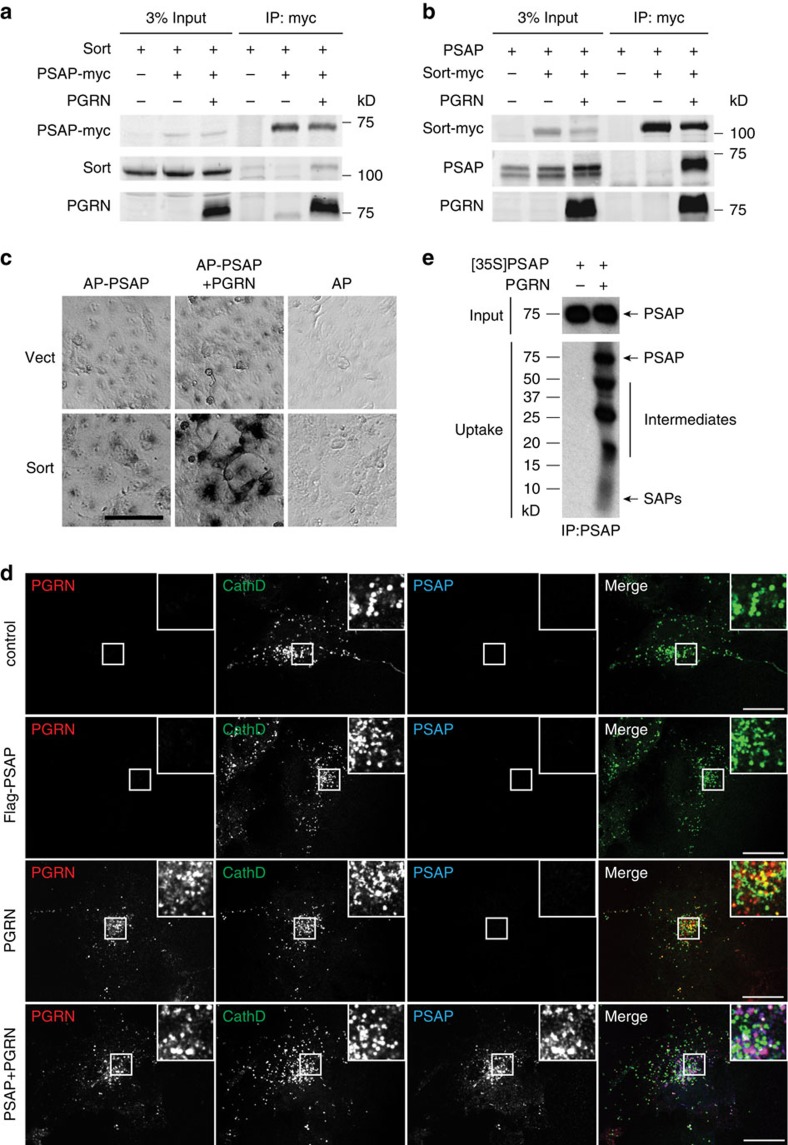
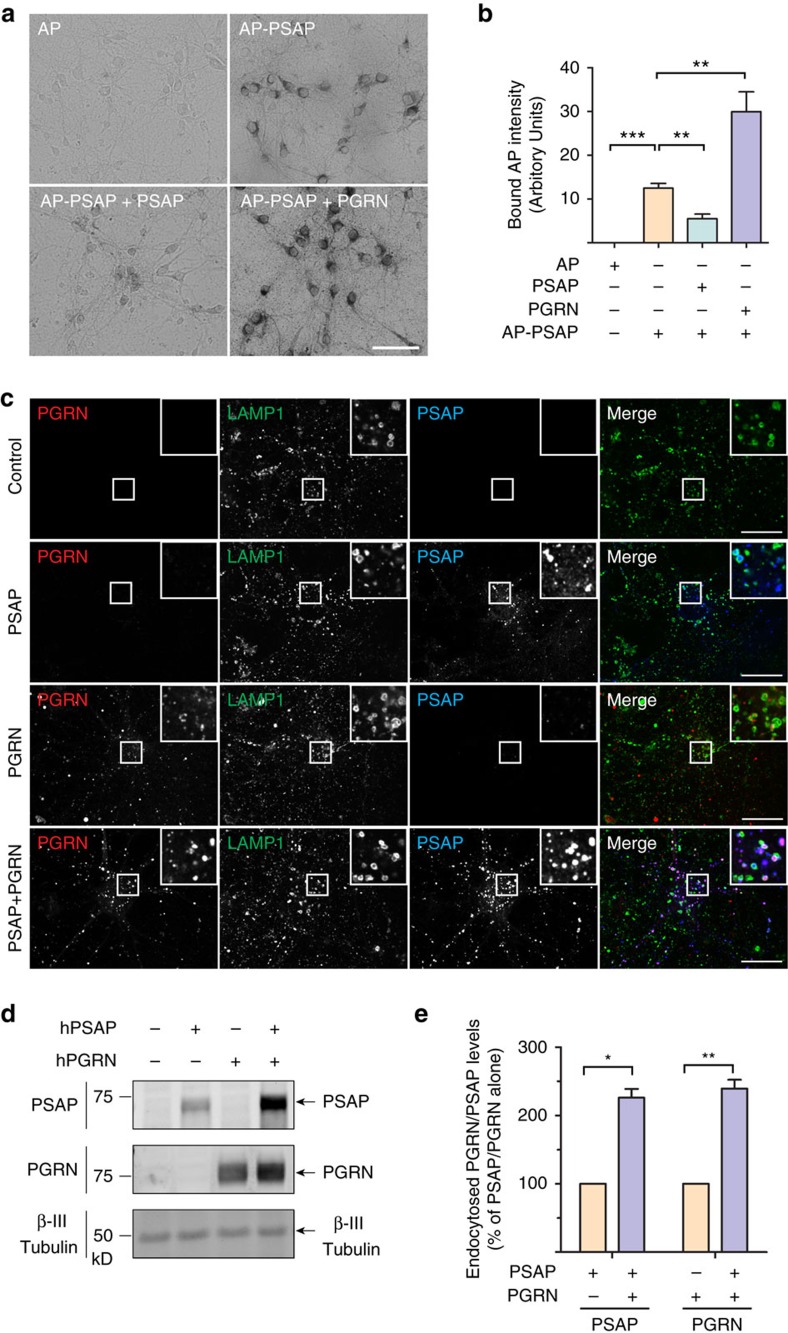
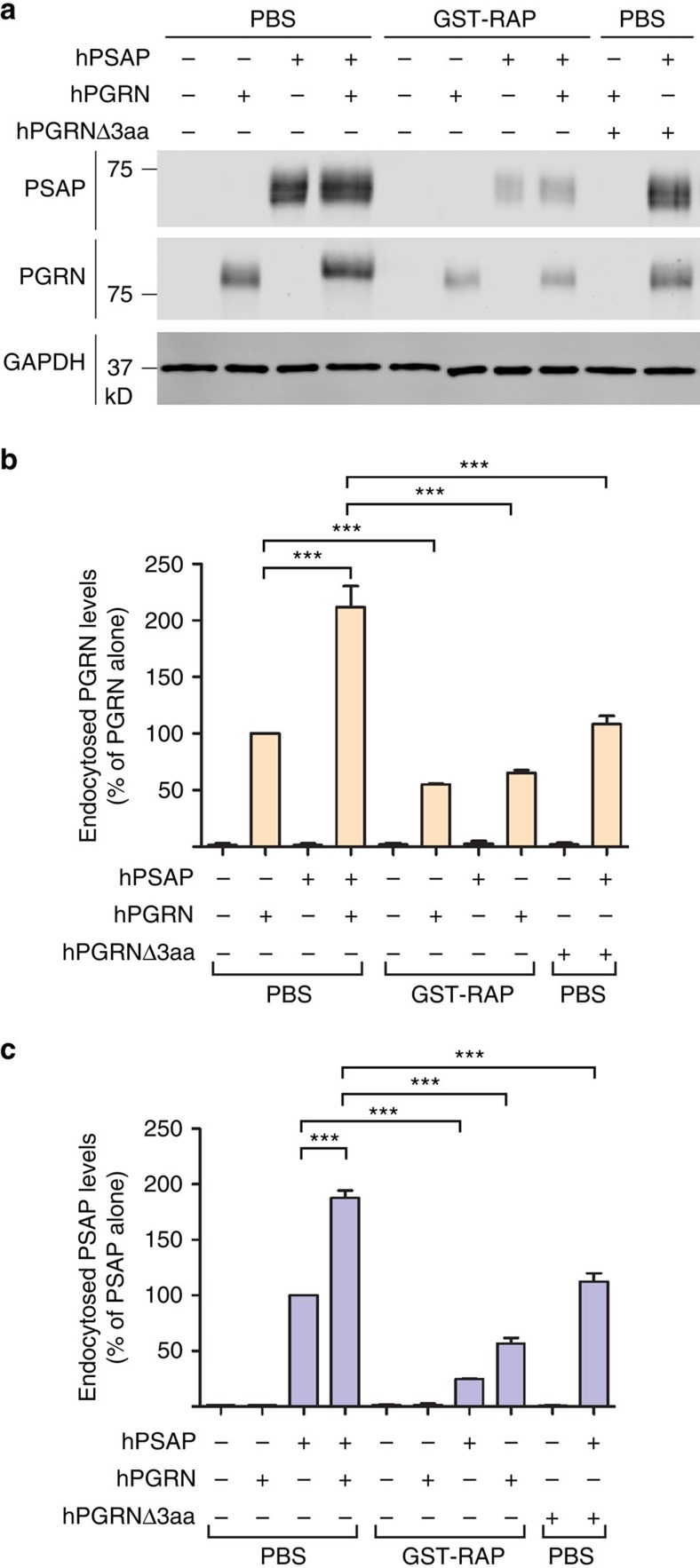
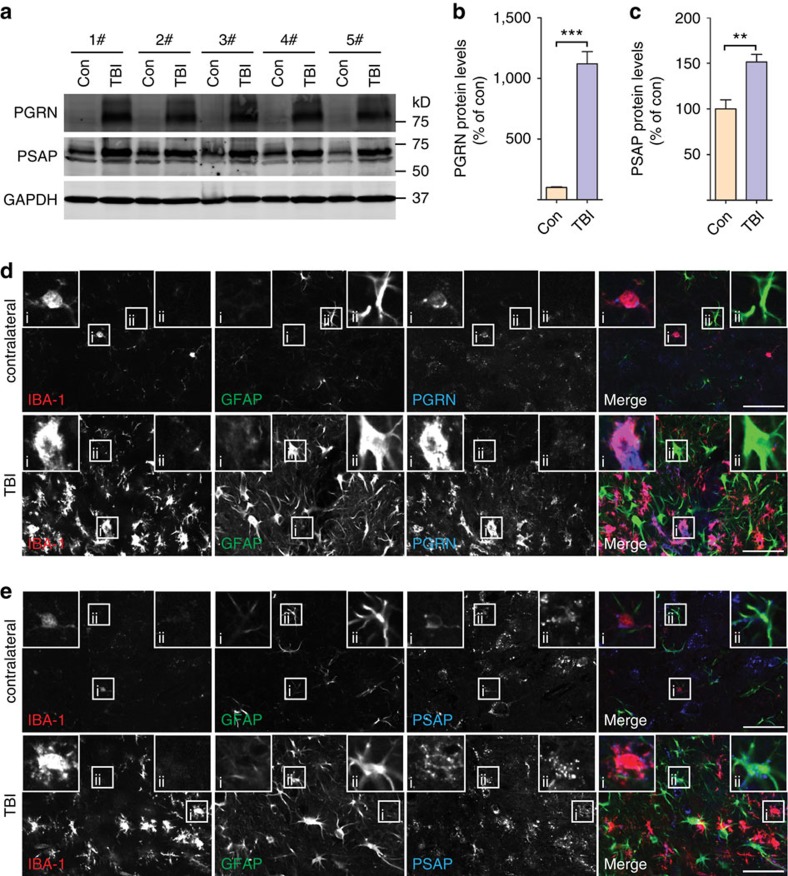
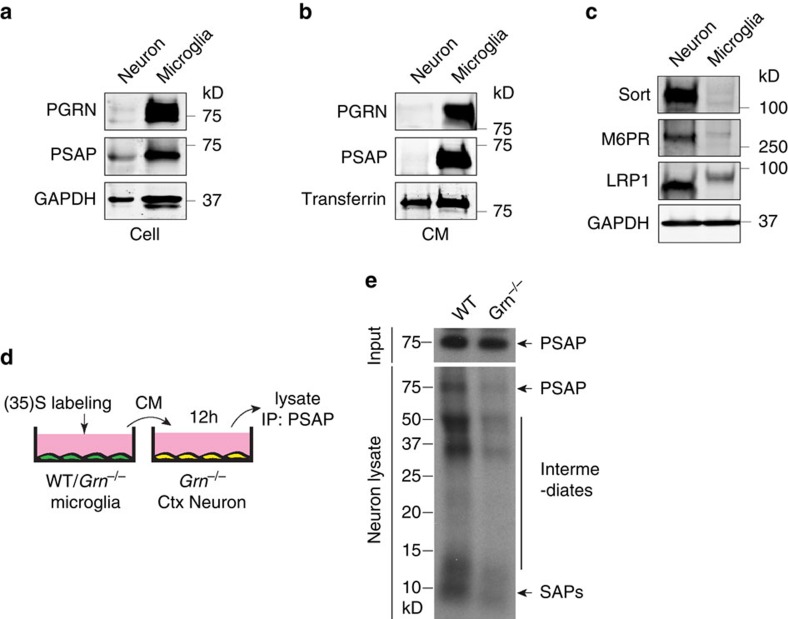
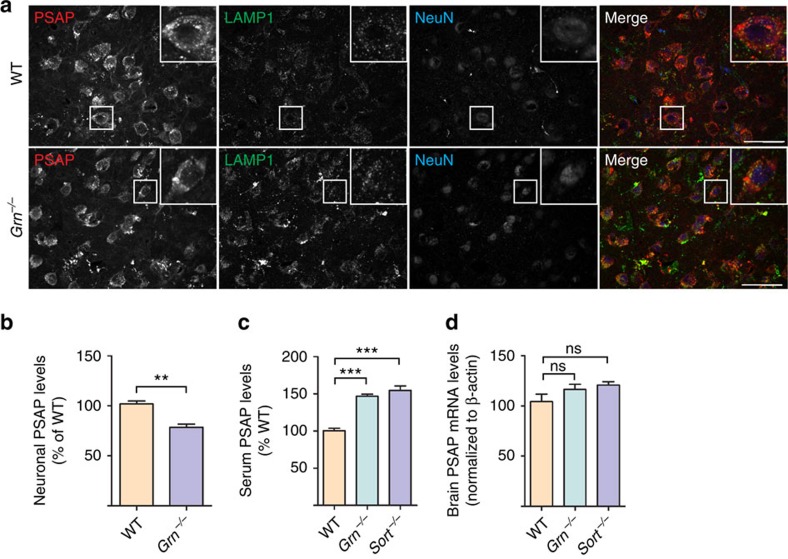
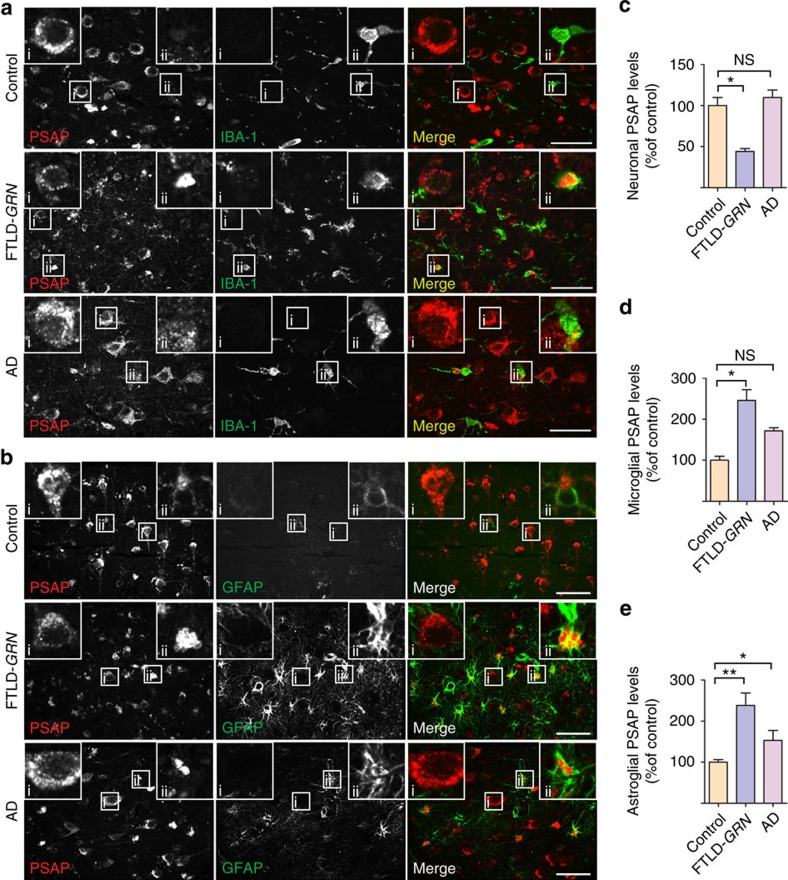
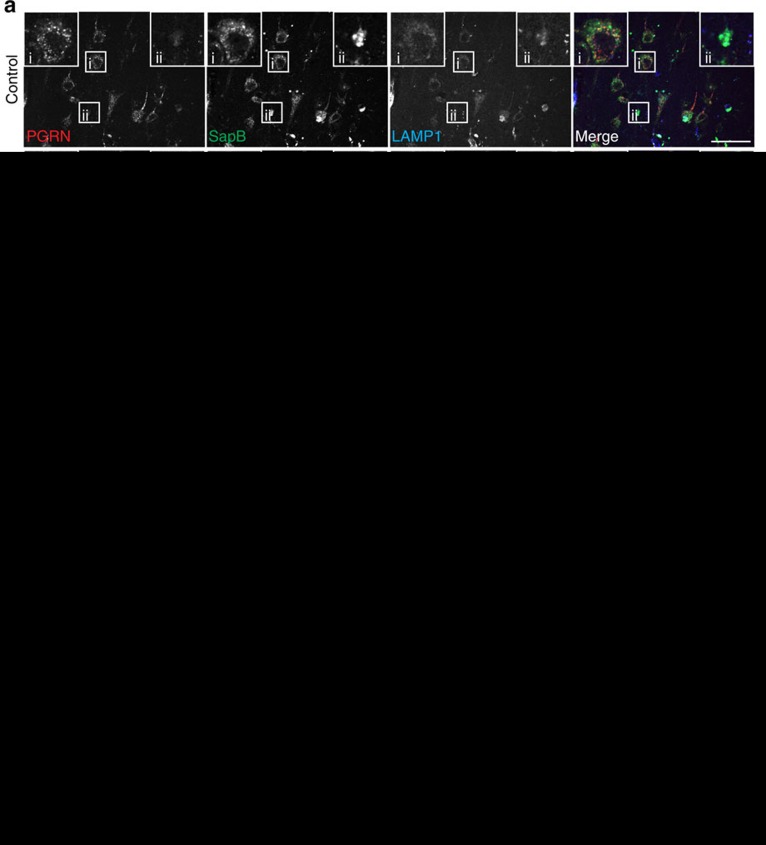
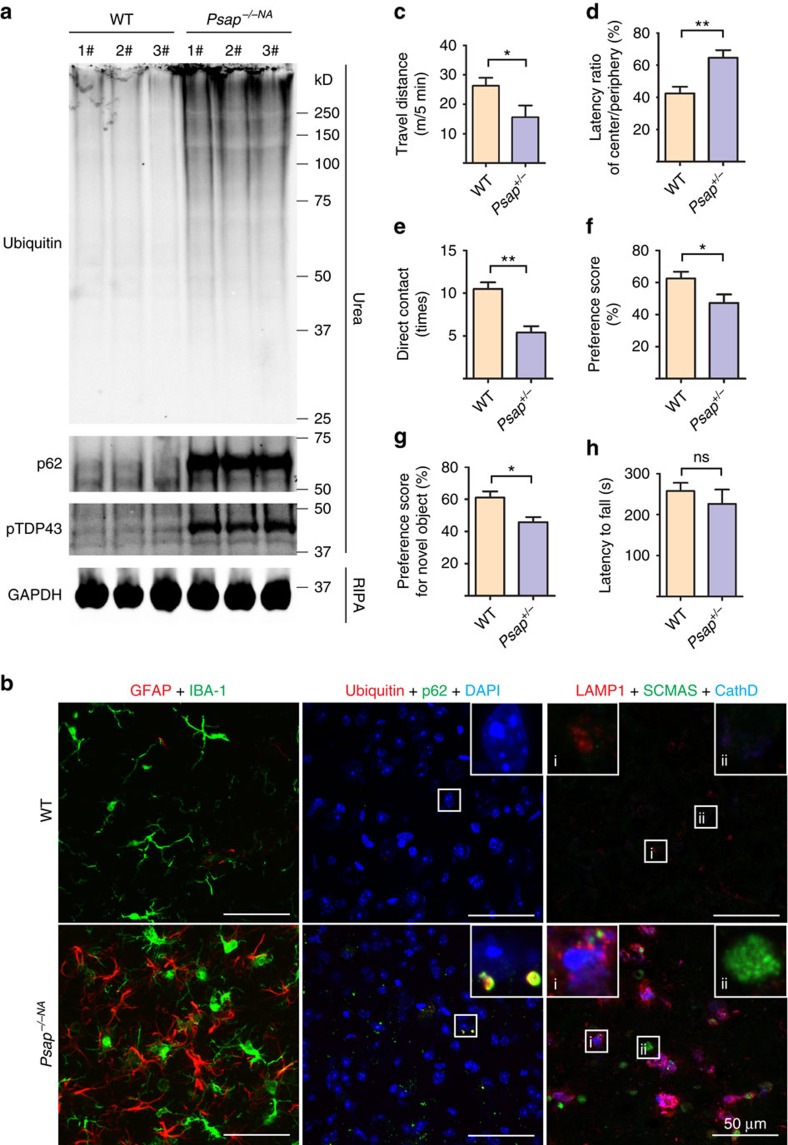
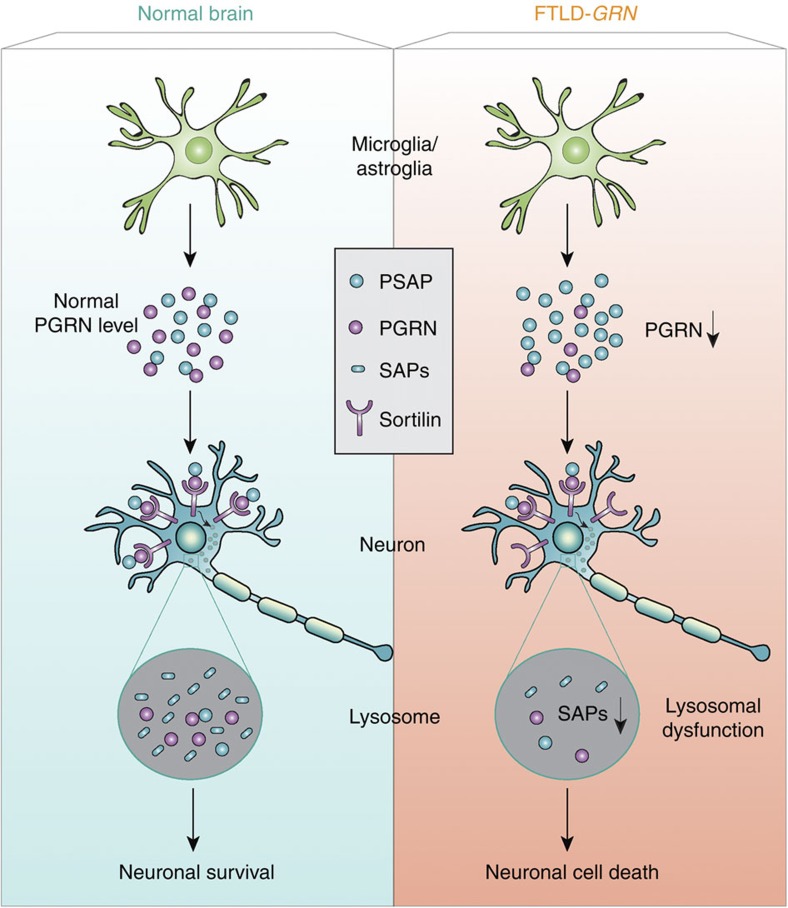
Haploinsufficiency of progranulin (PGRN) due to mutations in the granulin (GRN) gene causes frontotemporal lobar degeneration (FTLD), and complete loss of PGRN leads to a lysosomal storage disorder, neuronal ceroid lipofuscinosis (NCL). Accumulating evidence suggests that PGRN is essential for proper lysosomal function, but the precise mechanisms involved are not known. Here, we show that PGRN facilitates neuronal uptake and lysosomal delivery of prosaposin (PSAP), the precursor of saposin peptides that are essential for lysosomal glycosphingolipid degradation. We found reduced levels of PSAP in neurons both in mice deficient in PGRN and in human samples from FTLD patients due to GRN mutations. Furthermore, mice with reduced PSAP expression demonstrated FTLD-like pathology and behavioural changes. Thus, our data demonstrate a role of PGRN in PSAP lysosomal trafficking and suggest that impaired lysosomal trafficking of PSAP is an underlying disease mechanism for NCL and FTLD due to GRN mutations.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Early lysosomal maturation deficits in microglia triggers enhanced lysosomal activity in other brain cells of progranulin knockout mice.Mol Neurodegener. 2018 Sep 4;13(1):48. doi: 10.1186/s13024-018-0281-5. Mol Neurodegener. 2018. PMID: 30180904 Free PMC article.
-
Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin.J Cell Biol. 2015 Sep 14;210(6):991-1002. doi: 10.1083/jcb.201502029. J Cell Biol. 2015. PMID: 26370502 Free PMC article.
-
Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis.Acta Neuropathol. 2014;127(6):845-60. doi: 10.1007/s00401-014-1262-6. Epub 2014 Mar 12. Acta Neuropathol. 2014. PMID: 24619111
-
Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia.Trends Cell Biol. 2023 Apr;33(4):324-339. doi: 10.1016/j.tcb.2022.09.006. Epub 2022 Oct 13. Trends Cell Biol. 2023. PMID: 36244875 Review.
-
The lysosomal function of progranulin, a guardian against neurodegeneration.Acta Neuropathol. 2018 Jul;136(1):1-17. doi: 10.1007/s00401-018-1861-8. Epub 2018 May 9. Acta Neuropathol. 2018. PMID: 29744576 Free PMC article. Review.
Cited by
-
Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations.Acta Neuropathol Commun. 2020 Oct 7;8(1):163. doi: 10.1186/s40478-020-01037-x. Acta Neuropathol Commun. 2020. PMID: 33028409 Free PMC article.
-
Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic.Cell. 2021 Sep 2;184(18):4651-4668.e25. doi: 10.1016/j.cell.2021.08.002. Epub 2021 Aug 26. Cell. 2021. PMID: 34450028 Free PMC article.
-
Progranulin prevents regulatory NK cell cytotoxicity against antiviral T cells.JCI Insight. 2019 Sep 5;4(17):e129856. doi: 10.1172/jci.insight.129856. JCI Insight. 2019. PMID: 31484831 Free PMC article.
-
Regulation of extracellular progranulin in medial prefrontal cortex.Neurobiol Dis. 2023 Nov;188:106326. doi: 10.1016/j.nbd.2023.106326. Epub 2023 Oct 12. Neurobiol Dis. 2023. PMID: 37838007 Free PMC article.
-
Characterization of lysosomal proteins Progranulin and Prosaposin and their interactions in Alzheimer's disease and aged brains: increased levels correlate with neuropathology.Acta Neuropathol Commun. 2019 Dec 21;7(1):215. doi: 10.1186/s40478-019-0862-8. Acta Neuropathol Commun. 2019. PMID: 31864418 Free PMC article.
References
-
- Neary D. et al.. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554 (1998). - PubMed
-
- Neumann M. et al.. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). - PubMed
-
- Arai T. et al.. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 (2006). - PubMed
-
- Baker M. et al.. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 (2006). - PubMed
-
- Cruts M. et al.. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
