

Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells
- PMID: 28542559
- PMCID: PMC5439952
- DOI: 10.1371/journal.pone.0178278
Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells
Abstract
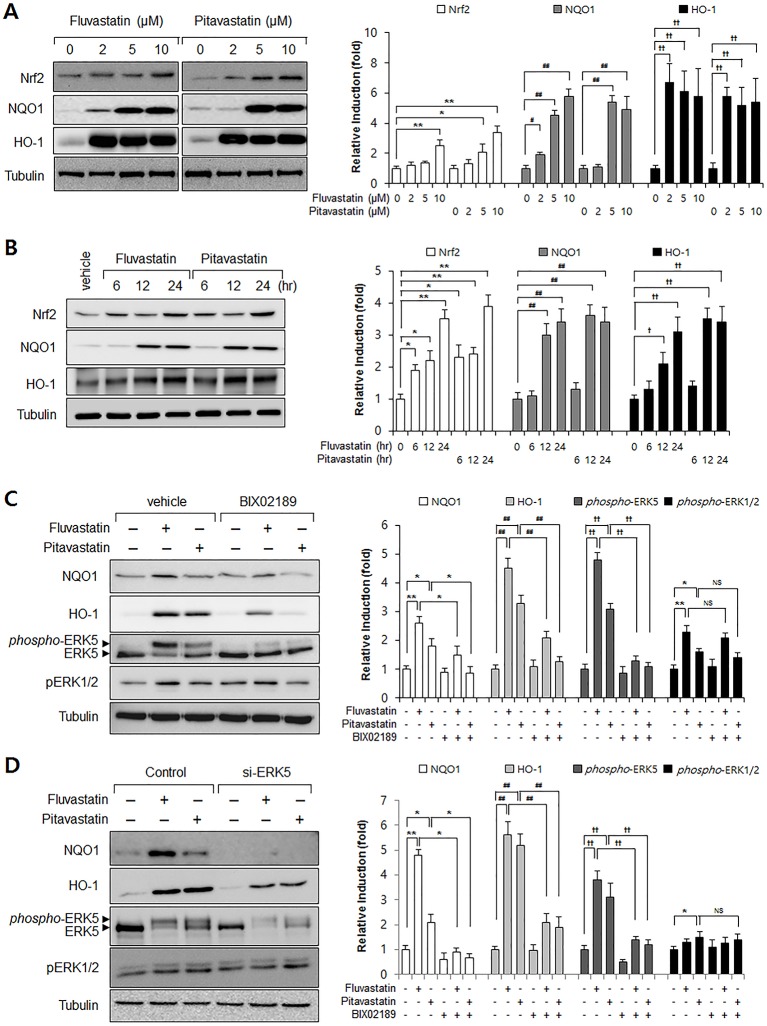
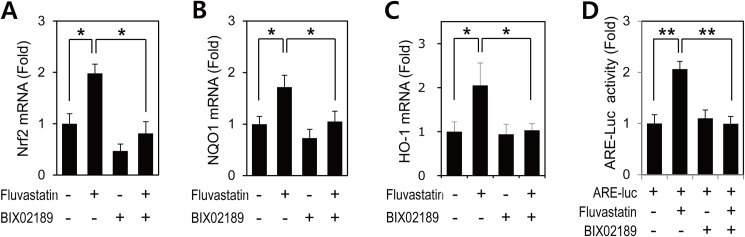
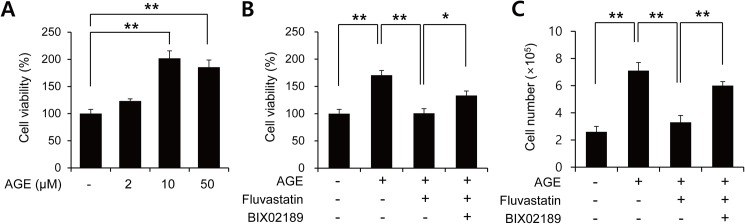
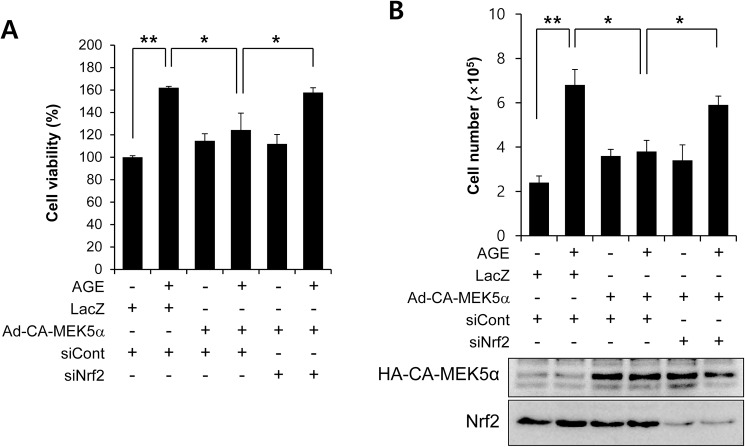
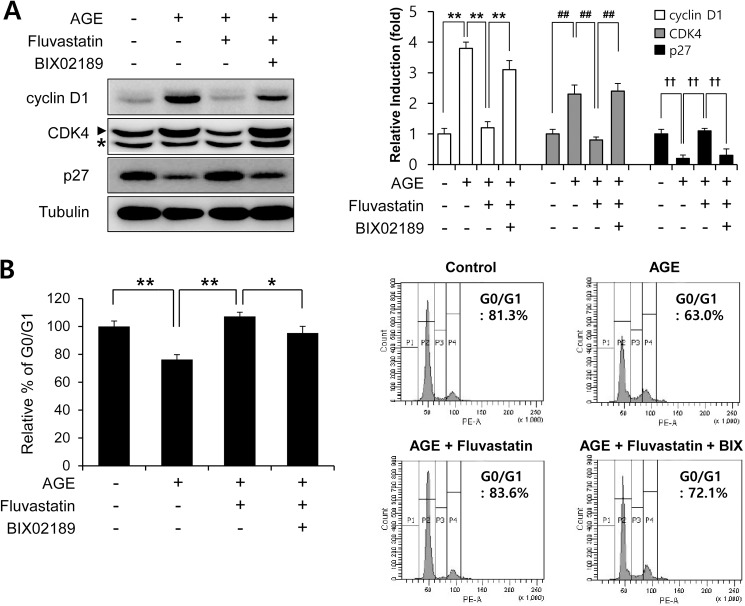
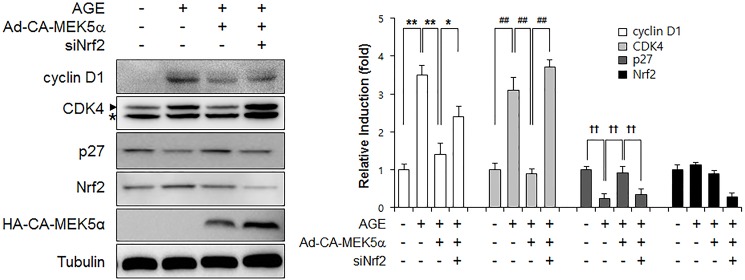
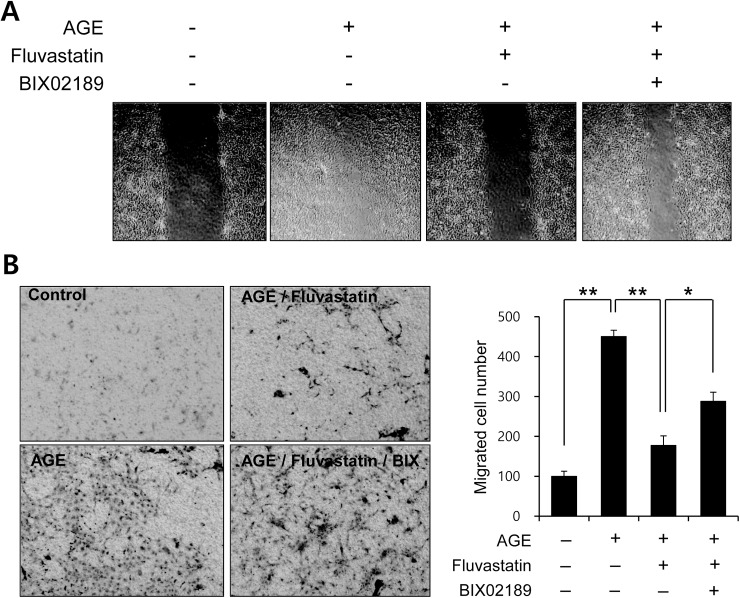
Advanced glycation endproduct (AGE)-induced vascular smooth muscle cell (VSMC) proliferation and reactive oxygen species (ROS) production are emerging as important mechanisms of diabetic vasculopathy, but little is known about the molecular mechanism responsible for the antioxidative effects of statins on AGEs. It has been reported that statins exert pleiotropic effects on the cardiovascular system due to decreases in AGE-induced cell proliferation, migration, and vascular inflammation. Thus, in the present study, the authors investigated the molecular mechanism by which statins decrease AGE-induced cell proliferation and VSMC migration. In cultured VSMCs, statins upregulated Nrf2-related antioxidant gene, NQO1 and HO-1, via an ERK5-dependent Nrf2 pathway. Inhibition of ERK5 by siRNA or BIX02189 (a specific ERK5 inhibitor) reduced the statin-induced upregulations of Nrf2, NQO1, and HO-1. Furthermore, fluvastatin was found to significantly increase ARE promoter activity through ERK5 signaling, and to inhibit AGE-induced VSMC proliferation and migration as determined by MTT assay, cell counting, FACS analysis, a wound scratch assay, and a migration chamber assay. In addition, AGE-induced proliferation was diminished in the presence of Ad-CA-MEK5α encoding a constitutively active mutant form of MEK5α (an upstream kinase of ERK5), whereas depletion of Nrf2 restored statin-mediated reduction of AGE-induced cell proliferation. Moreover, fluvastatin suppressed the protein expressions of cyclin D1 and Cdk4, but induced p27, and blocked VSMC proliferation by regulating cell cycle. These results suggest statin-induced activation of an ERK5-dependent Nrf2 pathway reduces VSMC proliferation and migration induced by AGEs, and that the ERK5-Nrf2 signal module be viewed as a potential therapeutic target of vasculopathy in patients with diabetes and complications of the disease.
Conflict of interest statement
Figures
Similar articles
-
Activation of M3AChR (Type 3 Muscarinic Acetylcholine Receptor) and Nrf2 (Nuclear Factor Erythroid 2-Related Factor 2) Signaling by Choline Alleviates Vascular Smooth Muscle Cell Phenotypic Switching and Vascular Remodeling.Arterioscler Thromb Vasc Biol. 2020 Nov;40(11):2649-2664. doi: 10.1161/ATVBAHA.120.315146. Epub 2020 Sep 17. Arterioscler Thromb Vasc Biol. 2020. PMID: 32938216
-
Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia.Arterioscler Thromb Vasc Biol. 2013 Apr;33(4):760-8. doi: 10.1161/ATVBAHA.112.300614. Epub 2013 Feb 14. Arterioscler Thromb Vasc Biol. 2013. PMID: 23413426
-
Advanced glycation end products promote the proliferation and migration of primary rat vascular smooth muscle cells via the upregulation of BAG3.Int J Mol Med. 2017 May;39(5):1242-1254. doi: 10.3892/ijmm.2017.2938. Epub 2017 Mar 28. Int J Mol Med. 2017. PMID: 28350077 Free PMC article.
-
Role of big mitogen-activated protein kinase 1 (BMK1) / extracellular signal-regulated kinase 5 (ERK5) in the pathogenesis and progression of atherosclerosis.J Pharmacol Sci. 2012;120(4):259-63. doi: 10.1254/jphs.12r11cp. Epub 2012 Nov 20. J Pharmacol Sci. 2012. PMID: 23165802 Review.
-
New possible pharmacological targets for statins and ezetimibe.Biomed Pharmacother. 2020 Sep;129:110388. doi: 10.1016/j.biopha.2020.110388. Epub 2020 Jun 16. Biomed Pharmacother. 2020. PMID: 32559626 Review.
Cited by
-
Antioxidant Effects of Statins by Modulating Nrf2 and Nrf2/HO-1 Signaling in Different Diseases.J Clin Med. 2022 Feb 27;11(5):1313. doi: 10.3390/jcm11051313. J Clin Med. 2022. PMID: 35268403 Free PMC article. Review.
-
Methylglyoxal in Cardiometabolic Disorders: Routes Leading to Pathology Counterbalanced by Treatment Strategies.Molecules. 2023 Nov 24;28(23):7742. doi: 10.3390/molecules28237742. Molecules. 2023. PMID: 38067472 Free PMC article. Review.
-
Role of advanced glycation end products on vascular smooth muscle cells under diabetic atherosclerosis.Front Endocrinol (Lausanne). 2022 Aug 31;13:983723. doi: 10.3389/fendo.2022.983723. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36120471 Free PMC article. Review.
-
Fluvastatin inhibits advanced glycation end products-induced proliferation, migration, and extracellular matrix accumulation in vascular smooth muscle cells by targeting connective tissue growth factor.Korean J Physiol Pharmacol. 2018 Mar;22(2):193-201. doi: 10.4196/kjpp.2018.22.2.193. Epub 2018 Feb 23. Korean J Physiol Pharmacol. 2018. PMID: 29520172 Free PMC article.
-
Senescent Phenotype Induced by p90RSK-NRF2 Signaling Sensitizes Monocytes and Macrophages to Oxidative Stress in HIV-Positive Individuals.Circulation. 2019 Feb 26;139(9):1199-1216. doi: 10.1161/CIRCULATIONAHA.118.036232. Circulation. 2019. PMID: 30586719 Free PMC article. Clinical Trial.
References
-
- Maron DJ. The epidemiology of low levels of high-density lipoprotein cholesterol in patients with and without coronary artery disease. Am J Cardiol. 2000;86(12A):11L–4L. - PubMed
-
- Pedersen TR, Wilhelmsen L, Faergeman O, Strandberg TE, Thorgeirsson G, Troedsson L, et al. Follow-up study of patients randomized in the Scandinavian simvastatin survival study (4S) of cholesterol lowering. Am J Cardiol. 2000;86(3):257–62. - PubMed
-
- Zhou Z, Wang K, Penn MS, Marso SP, Lauer MA, Forudi F, et al. Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation. 2003;107(17):2238–43. doi: 10.1161/01.CIR.0000063577.32819.23 - DOI - PubMed
-
- Duckers HJ, Boehm M, True AL, Yet SF, San H, Park JL, et al. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat Med. 2001;7(6):693–8. doi: 10.1038/89068 - DOI - PubMed
-
- Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18(6):753–60. doi: 10.1016/j.cellsig.2005.11.003 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
