

Incidence of Atrophic Lesions in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 5
- PMID: 28542697
- PMCID: PMC5710205
- DOI: 10.1001/jamaophthalmol.2017.1121
Incidence of Atrophic Lesions in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 5
Abstract
Importance: Outcome measures that are sensitive to disease progression are needed as clinical end points for future treatment trials in Stargardt disease.
Objective: To examine the incidence of atrophic lesions of the retinal pigment epithelium in patients with Stargardt disease as determined by fundus autofluorescence imaging.
Design, setting, and participants: In this retrospective multicenter cohort study, 217 patients 6 years and older at baseline at tertiary referral centers in Europe, the United States, and the United Kingdom who were harboring disease-causing variants in the adenosine triphosphate (ATP)-binding cassette subfamily A member 4 (ABCA4) gene and who met the following criteria were enrolled: (1) at least 1 well-demarcated area of atrophy with a minimum diameter of 300 µm, with the total area of all atrophic lesions being less than or equal to 12 mm2 in at least 1 eye at the most recent visit, and (2) fundus autofluorescence images for at least 2 visits with a minimum of 6 months between at least 2 visits. Data were collected between August 22, 2013, and December 12, 2014. Data analysis was performed from March 15, 2015, through January 31, 2017.
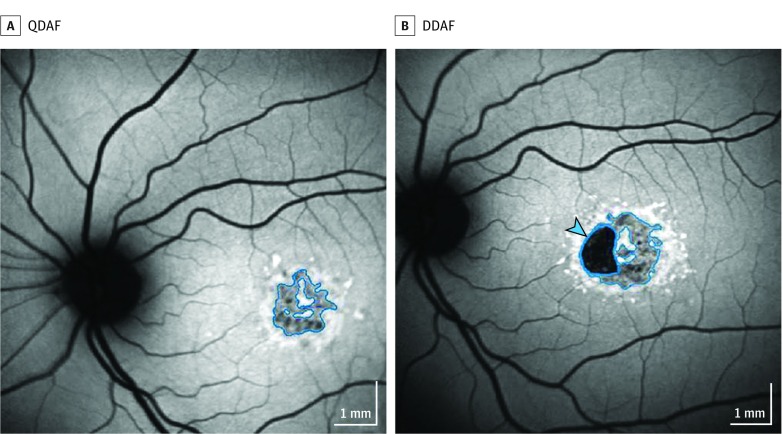
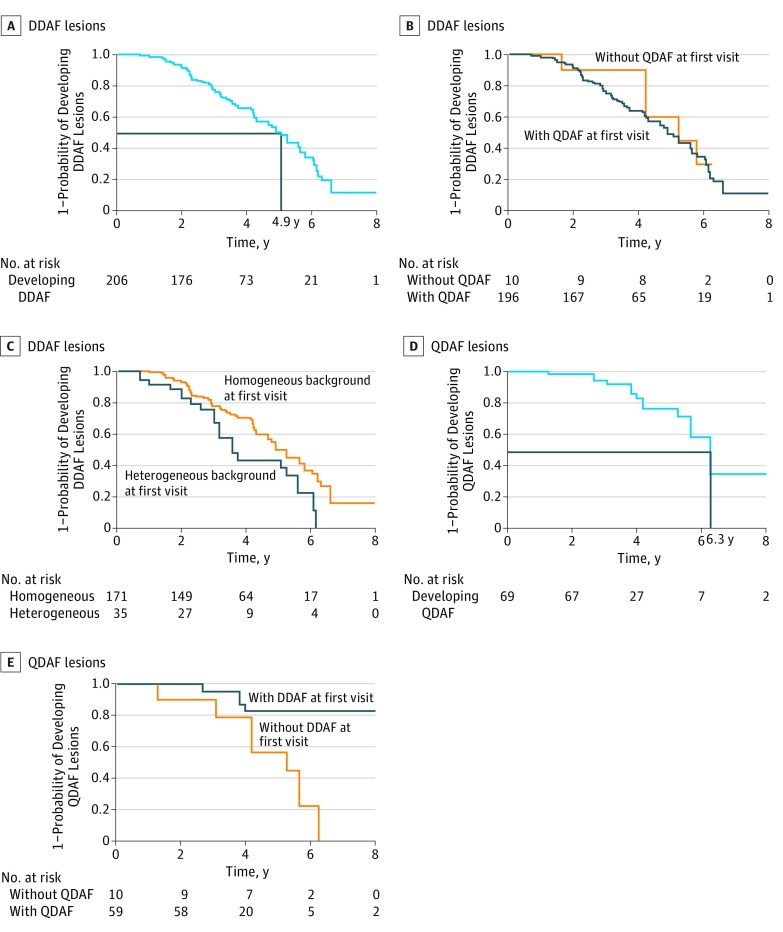
Exposures: Images were evaluated by staff at a central reading center. Areas of definitely decreased autofluorescence (DDAF) and questionably decreased autofluorescence (QDAF) were outlined and quantified. Lesion-free survival rates were estimated using Kaplan-Meier survival curves.
Main outcomes and measures: Incidence of atrophic lesions as determined by fundus autofluorescence.
Results: The 217 patients (mean [SD] age, 21.8 [13.3] years; 127 female [57.5%]; 148 white [68.2%]) contributed 390 eyes for which the mean (SD) follow-up time was 3.9 (1.6) years (range, 0.7-12.1 years). Among eyes without DDAF at first visit, the median time to develop a DDAF lesion was 4.9 years (95% CI, 4.3-5.6 years). Among eyes without QDAF, the median time to develop a QDAF lesion was 6.3 years (95% CI, 5.6-9.7 years). Eyes with a lesion of DDAF at the first visit were less likely to develop a QDAF lesion compared with eyes without a lesion of DDAF (hazard ratio, 0.19; 95% CI, 0.05-0.70; P = .01).
Conclusions and relevance: An estimated 50% of the eyes without DDAF at first visit will develop the lesion in less than 5 years, suggesting that incidence of DDAF could serve as an outcome measure for treatment trials.
Conflict of interest statement
Figures
Comment in
-
The Importance of Outcome Measure Research in Stargardt Disease.JAMA Ophthalmol. 2017 Jul 1;135(7):704-705. doi: 10.1001/jamaophthalmol.2017.1544. JAMA Ophthalmol. 2017. PMID: 28542669 Free PMC article. No abstract available.
References
-
- Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236-246. - PubMed
-
- Strauss RW, Ho A, Muñoz B, et al. ; Progression of Stargardt Disease Study Group . The natural history of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) studies: design and baseline characteristics: ProgStar report No. 1. Ophthalmology. 2016;123(4):817-828. - PubMed
-
- Scholl HP, Strauss RW, Singh MS, et al. Emerging therapies for inherited retinal degeneration. Sci Transl Med. 2016;8(368):368rv6. - PubMed
-
- Csaky KG, Richman EA, Ferris FL III. Report from the NEI/FDA Ophthalmic Clinical Trial Design and Endpoints Symposium. Invest Ophthalmol Vis Sci. 2008;49(2):479-489. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
