

Modulatory features of the novel spider toxin μ-TRTX-Df1a isolated from the venom of the spider Davus fasciatus
- PMID: 28542706
- PMCID: PMC5513869
- DOI: 10.1111/bph.13865
Modulatory features of the novel spider toxin μ-TRTX-Df1a isolated from the venom of the spider Davus fasciatus
Abstract
Background and purpose: Naturally occurring dysfunction of voltage-gated sodium (NaV ) channels results in complex disorders such as chronic pain, making these channels an attractive target for new therapies. In the pursuit of novel NaV modulators, we investigated spider venoms for new inhibitors of NaV channels.
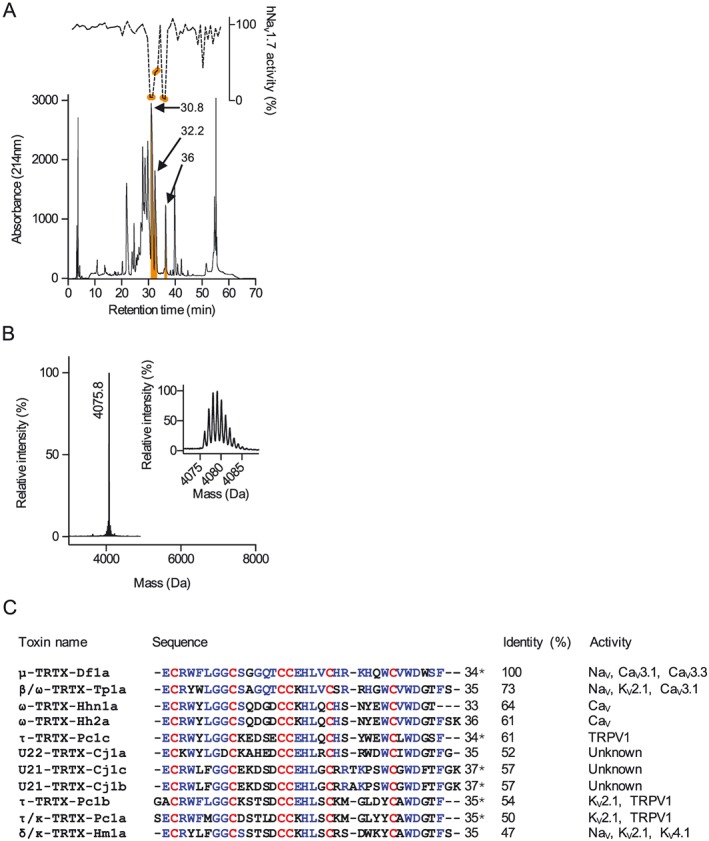
Experimental approach: We used high-throughput screens to identify a NaV modulator in venom of the spider Davus fasciatus. Further characterization of this venom peptide was undertaken using fluorescent and electrophysiological assays, molecular modelling and a rodent pain model.
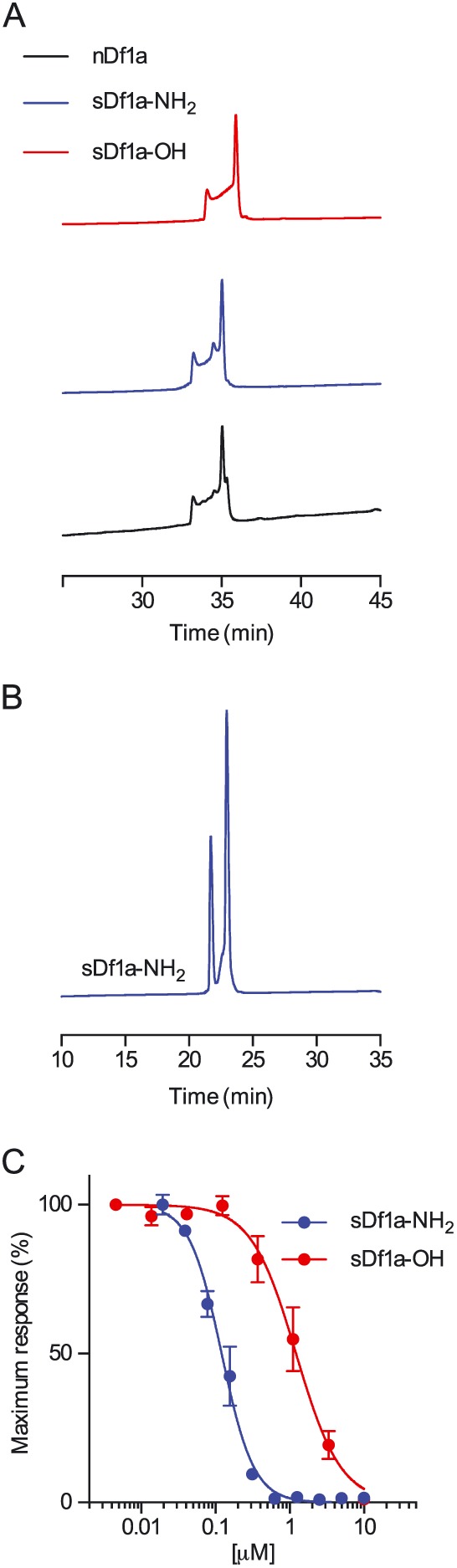
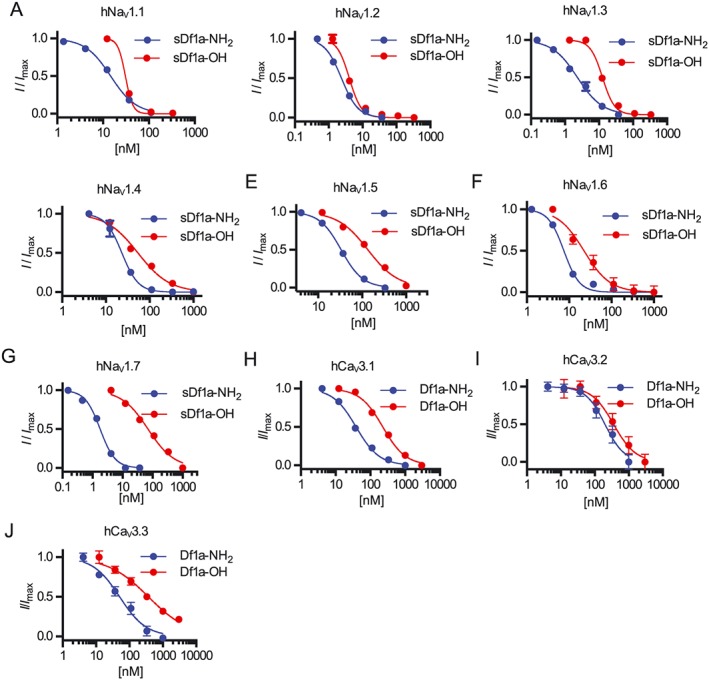
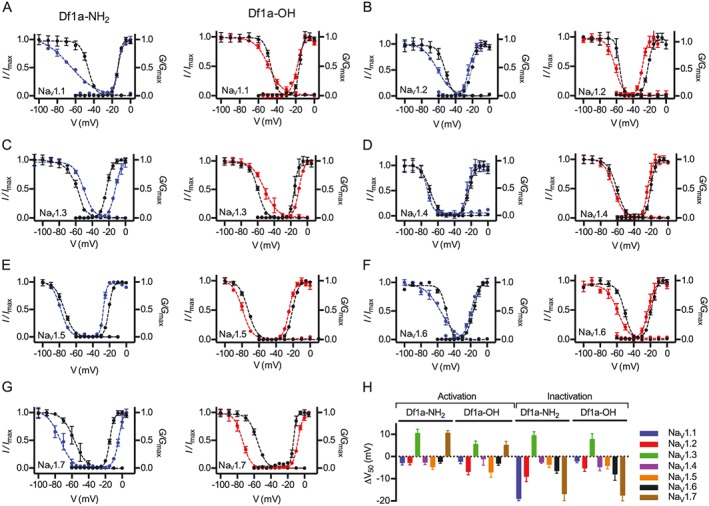
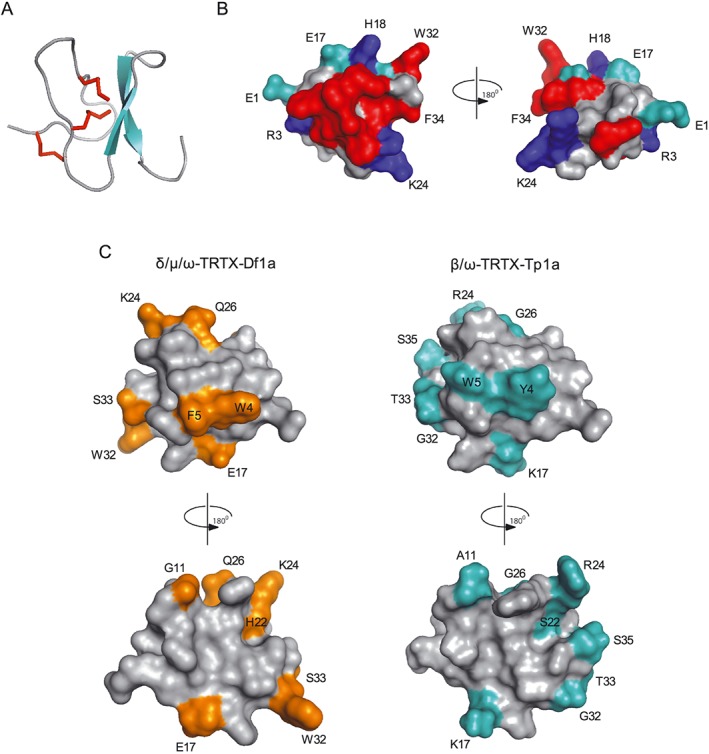
Key results: We identified a potent NaV inhibitor named μ-TRTX-Df1a. This 34-residue peptide fully inhibited responses mediated by NaV 1.7 endogenously expressed in SH-SY5Y cells. Df1a also inhibited voltage-gated calcium (CaV 3) currents but had no activity against the voltage-gated potassium (KV 2) channel. The modelled structure of Df1a, which contains an inhibitor cystine knot motif, is reminiscent of the NaV channel toxin ProTx-I. Electrophysiology revealed that Df1a inhibits all NaV subtypes tested (hNaV 1.1-1.7). Df1a also slowed fast inactivation of NaV 1.1, NaV 1.3 and NaV 1.5 and modified the voltage-dependence of activation and inactivation of most of the NaV subtypes. Df1a preferentially binds to the domain II voltage-sensor and has additional interactions with the voltage sensors domains III and IV, which probably explains its modulatory features. Df1a was analgesic in vivo, reversing the spontaneous pain behaviours induced by the NaV activator OD1.
Conclusion and implications: μ-TRTX-Df1a shows potential as a new molecule for the development of drugs to treat pain disorders mediated by voltage-gated ion channels.
© 2017 The British Pharmacological Society.
Figures
Similar articles
-
Identification and Characterization of ProTx-III [μ-TRTX-Tp1a], a New Voltage-Gated Sodium Channel Inhibitor from Venom of the Tarantula Thrixopelma pruriens.Mol Pharmacol. 2015 Aug;88(2):291-303. doi: 10.1124/mol.115.098178. Epub 2015 May 15. Mol Pharmacol. 2015. PMID: 25979003
-
Seven novel modulators of the analgesic target NaV 1.7 uncovered using a high-throughput venom-based discovery approach.Br J Pharmacol. 2015 May;172(10):2445-58. doi: 10.1111/bph.13081. Epub 2015 Mar 4. Br J Pharmacol. 2015. PMID: 25754331 Free PMC article.
-
Evaluation of the Spider (Phlogiellus genus) Phlotoxin 1 and Synthetic Variants as Antinociceptive Drug Candidates.Toxins (Basel). 2019 Aug 22;11(9):484. doi: 10.3390/toxins11090484. Toxins (Basel). 2019. PMID: 31443554 Free PMC article.
-
Spider Knottin Pharmacology at Voltage-Gated Sodium Channels and Their Potential to Modulate Pain Pathways.Toxins (Basel). 2019 Oct 29;11(11):626. doi: 10.3390/toxins11110626. Toxins (Basel). 2019. PMID: 31671792 Free PMC article. Review.
-
Spider and scorpion knottins targeting voltage-gated sodium ion channels in pain signaling.Biochem Pharmacol. 2024 Sep;227:116465. doi: 10.1016/j.bcp.2024.116465. Epub 2024 Aug 3. Biochem Pharmacol. 2024. PMID: 39102991 Review.
Cited by
-
Unveiling the Pain Relief Potential: Harnessing Analgesic Peptides from Animal Venoms.Pharmaceutics. 2023 Dec 13;15(12):2766. doi: 10.3390/pharmaceutics15122766. Pharmaceutics. 2023. PMID: 38140106 Free PMC article. Review.
-
Venomous Noodles: The Evolution of Toxins in Nemertea through Positive Selection and Gene Duplication.Toxins (Basel). 2023 Nov 12;15(11):650. doi: 10.3390/toxins15110650. Toxins (Basel). 2023. PMID: 37999513 Free PMC article.
-
The NaV1.7 Channel Subtype as an Antinociceptive Target for Spider Toxins in Adult Dorsal Root Ganglia Neurons.Front Pharmacol. 2018 Sep 4;9:1000. doi: 10.3389/fphar.2018.01000. eCollection 2018. Front Pharmacol. 2018. PMID: 30233376 Free PMC article. Review.
-
Structure-Function and Therapeutic Potential of Spider Venom-Derived Cysteine Knot Peptides Targeting Sodium Channels.Front Pharmacol. 2019 Apr 11;10:366. doi: 10.3389/fphar.2019.00366. eCollection 2019. Front Pharmacol. 2019. PMID: 31031623 Free PMC article. Review.
-
Chemical synthesis of human trefoil factor 1 (TFF1) and its homodimer provides novel insights into their mechanisms of action.Chem Commun (Camb). 2020 Jun 14;56(47):6420-6423. doi: 10.1039/d0cc02321c. Epub 2020 May 11. Chem Commun (Camb). 2020. PMID: 32391824 Free PMC article.
References
-
- Arnold K, Bordoli L, Kopp J, Schwede T (2006). The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 22: 195–201. - PubMed
-
- Bosmans F, Rash L, Zhu S, Diochot S, Lazdunski M, Escoubas P et al. (2006). Four novel tarantula toxins as selective modulators of voltage‐gated sodium channel subtypes. Mol Pharmacol 69: 419–429. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
