

Spectroscopic evidence for the presence of a high-valent Fe(IV) species in the ferroxidase reaction of an archaeal ferritin
- PMID: 28542723
- PMCID: PMC5519934
- DOI: 10.1002/1873-3468.12697
Spectroscopic evidence for the presence of a high-valent Fe(IV) species in the ferroxidase reaction of an archaeal ferritin
Abstract
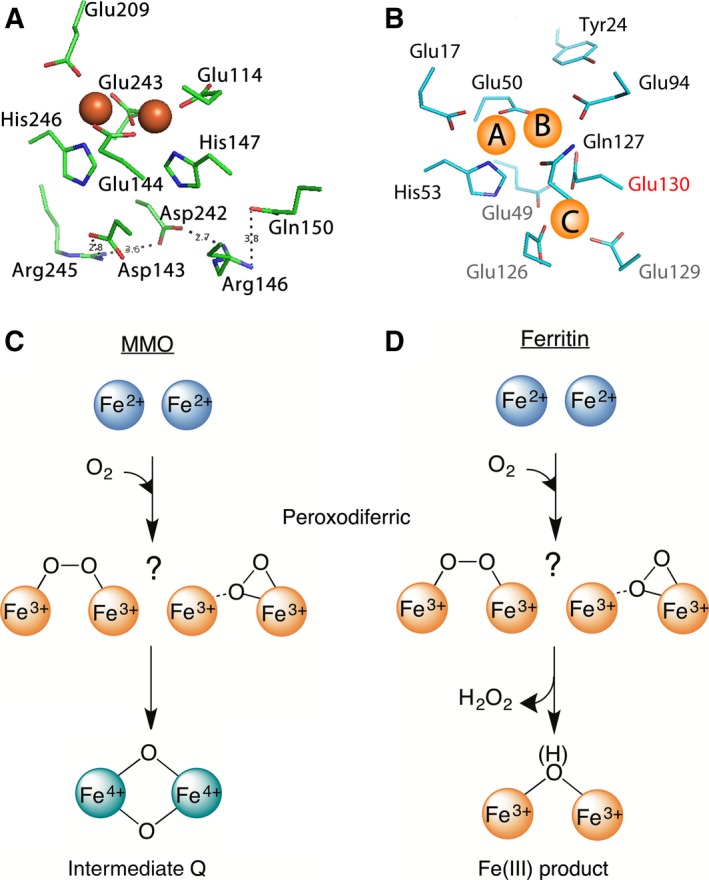
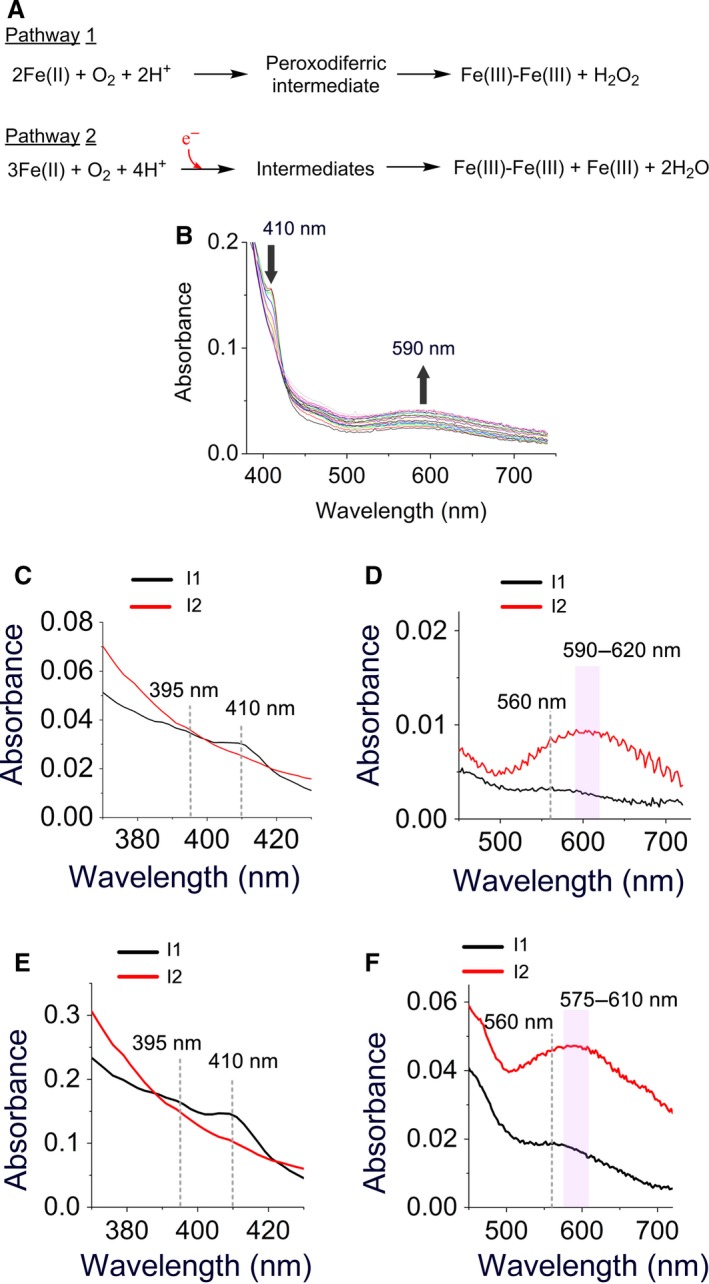
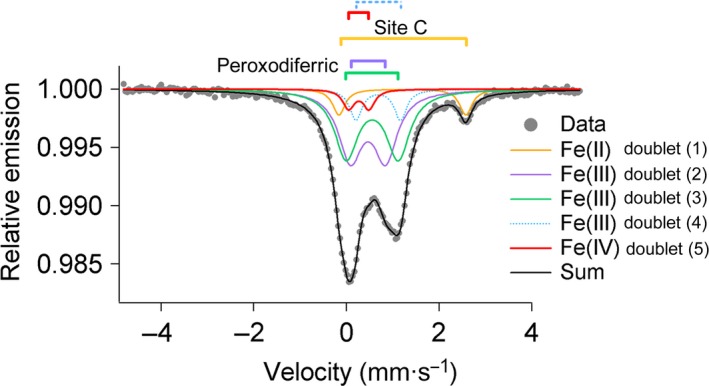
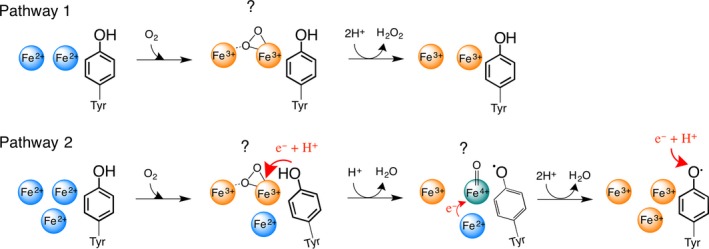
A high-valent Fe(IV) species is proposed to be generated from the decay of a peroxodiferric intermediate in the catalytic cycle at the di-iron cofactor center of dioxygen-activating enzymes such as methane monooxygenase. However, it is believed that this intermediate is not formed in the di-iron substrate site of ferritin, where oxidation of Fe(II) substrate to Fe(III) (the ferroxidase reaction) occurs also via a peroxodiferric intermediate. In opposition to this generally accepted view, here we present evidence for the occurrence of a high-valent Fe(IV) in the ferroxidase reaction of an archaeal ferritin, which is based on trapped intermediates obtained with the freeze-quench technique and combination of spectroscopic characterization. We hypothesize that a Fe(IV) intermediate catalyzes oxidation of excess Fe(II) nearby the ferroxidase center.
Keywords: Fe(IV); ferritin; ferroxidase; peroxodiferric; tyrosine radical.
© 2017 The Authors. FEBS Letters published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Figures
Similar articles
-
Self-assembly is prerequisite for catalysis of Fe(II) oxidation by catalytically active subunits of ferritin.J Biol Chem. 2015 Oct 30;290(44):26801-10. doi: 10.1074/jbc.M115.678375. Epub 2015 Sep 14. J Biol Chem. 2015. PMID: 26370076 Free PMC article.
-
Phosphate accelerates displacement of Fe(III) by Fe(II) in the ferroxidase center of Pyrococcus furiosus ferritin.FEBS Lett. 2013 Jan 16;587(2):220-5. doi: 10.1016/j.febslet.2012.11.029. Epub 2012 Dec 14. FEBS Lett. 2013. PMID: 23247211
-
The dinuclear iron-oxo ferroxidase center of Pyrococcus furiosus ferritin is a stable prosthetic group with unexpectedly high reduction potentials.FEBS Lett. 2005 Aug 29;579(21):4729-32. doi: 10.1016/j.febslet.2005.07.045. FEBS Lett. 2005. PMID: 16107254
-
Mechanisms of iron mineralization in ferritins: one size does not fit all.J Biol Inorg Chem. 2014 Aug;19(6):775-85. doi: 10.1007/s00775-014-1136-3. Epub 2014 Apr 19. J Biol Inorg Chem. 2014. PMID: 24748222 Review.
-
The iron redox and hydrolysis chemistry of the ferritins.Biochim Biophys Acta. 2010 Aug;1800(8):719-31. doi: 10.1016/j.bbagen.2010.03.021. Epub 2010 Apr 9. Biochim Biophys Acta. 2010. PMID: 20382203 Review.
Cited by
-
Encapsulated Ferritin-like Proteins: A Structural Perspective.Biomolecules. 2024 May 25;14(6):624. doi: 10.3390/biom14060624. Biomolecules. 2024. PMID: 38927029 Free PMC article. Review.
-
Iron Oxidation in Escherichia coli Bacterioferritin Ferroxidase Centre, a Site Designed to React Rapidly with H2O2 but Slowly with O2.Angew Chem Weinheim Bergstr Ger. 2021 Apr 6;133(15):8442-8450. doi: 10.1002/ange.202015964. Epub 2021 Mar 5. Angew Chem Weinheim Bergstr Ger. 2021. PMID: 38529354 Free PMC article.
-
Iron Oxidation in Escherichia coli Bacterioferritin Ferroxidase Centre, a Site Designed to React Rapidly with H2 O2 but Slowly with O2.Angew Chem Int Ed Engl. 2021 Apr 6;60(15):8361-8369. doi: 10.1002/anie.202015964. Angew Chem Int Ed Engl. 2021. PMID: 33482043 Free PMC article.
-
Structural characterization of the Myxococcus xanthus encapsulin and ferritin-like cargo system gives insight into its iron storage mechanism.Structure. 2022 Apr 7;30(4):551-563.e4. doi: 10.1016/j.str.2022.01.008. Epub 2022 Feb 11. Structure. 2022. PMID: 35150605 Free PMC article.
References
-
- Ebrahimi KH, Hagedoorn P‐L and Hagen WR (2015) Unity in the biochemistry of the iron‐storage proteins ferritin and bacterioferritin. Chem Rev 115, 295–326. - PubMed
-
- Ebrahimi KH, Bill E, Hagedoorn P‐L and Hagen WR (2012) The catalytic center of ferritin regulates iron storage via Fe (II)‐Fe (III) displacement. Nat Chem Biol 8, 941–948. - PubMed
-
- Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC and Huynh BH (1998) Direct spectroscopic and kinetic evidence for the involvement of a peroxodiferric intermediate during the ferroxidase reaction in fast ferritin mineralization. Biochemistry 37, 9871–9876. - PubMed
-
- Moënne‐Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH and Loehr TM (1999) The ferroxidase reaction of ferritin reveals a diferric μ‐1, 2 bridging peroxide intermediate in common with other O2‐activating non‐heme diiron proteins. Biochemistry 38, 5290–5295. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
