

Objective assessment of the evolutionary action equation for the fitness effect of missense mutations across CAGI-blinded contests
- PMID: 28544059
- PMCID: PMC5600169
- DOI: 10.1002/humu.23266
Objective assessment of the evolutionary action equation for the fitness effect of missense mutations across CAGI-blinded contests
Abstract
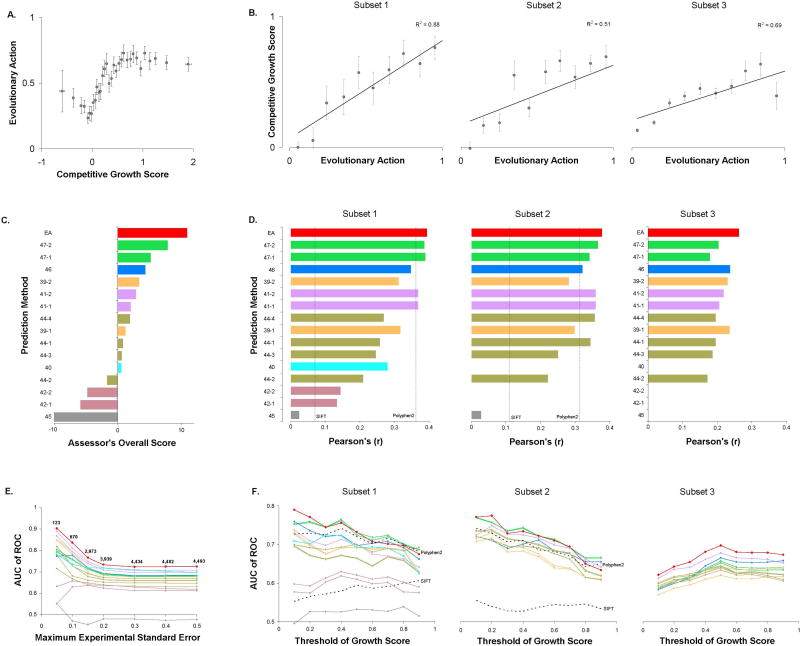
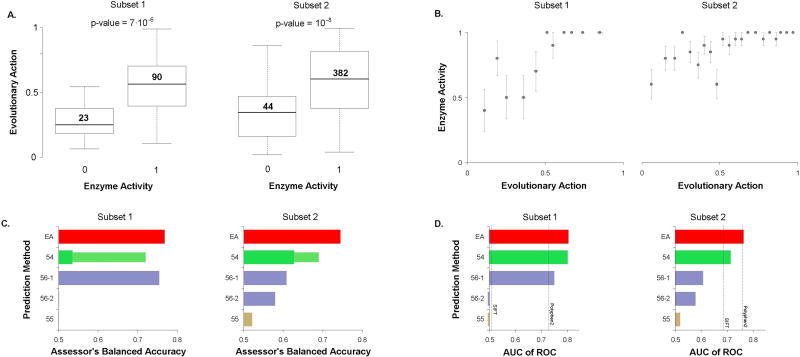
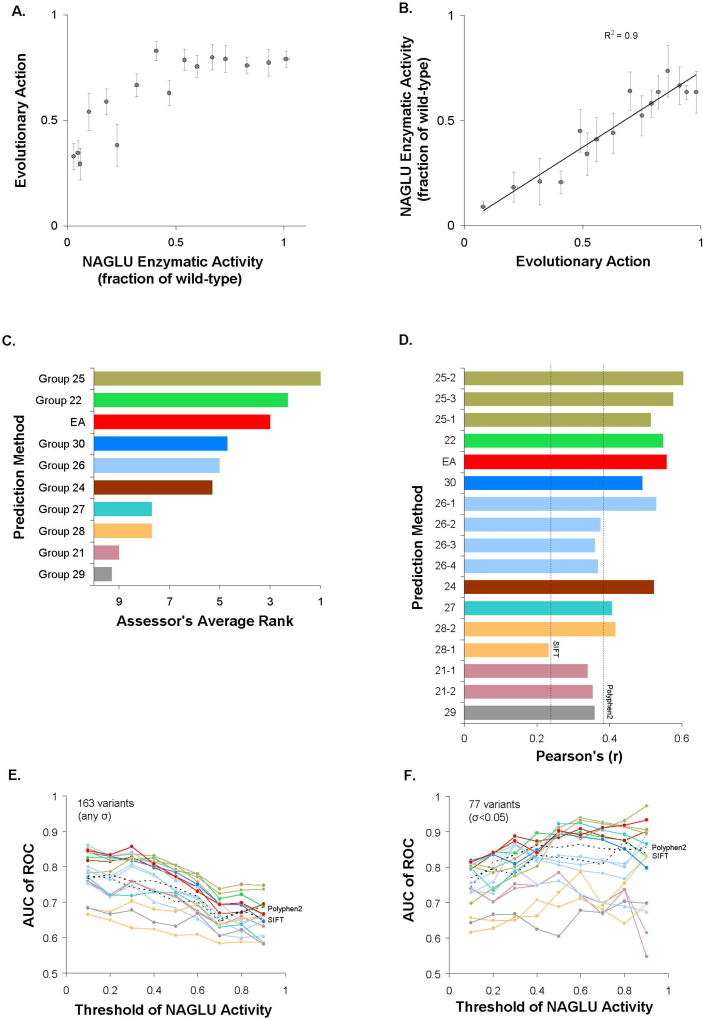
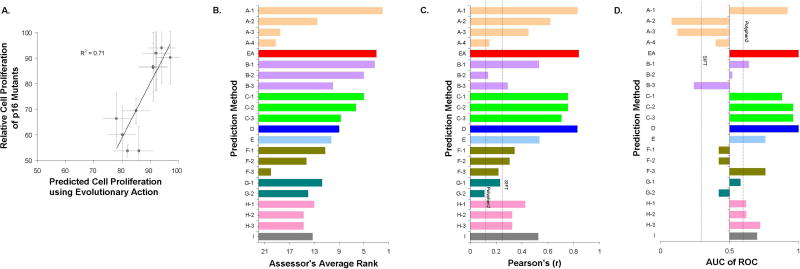
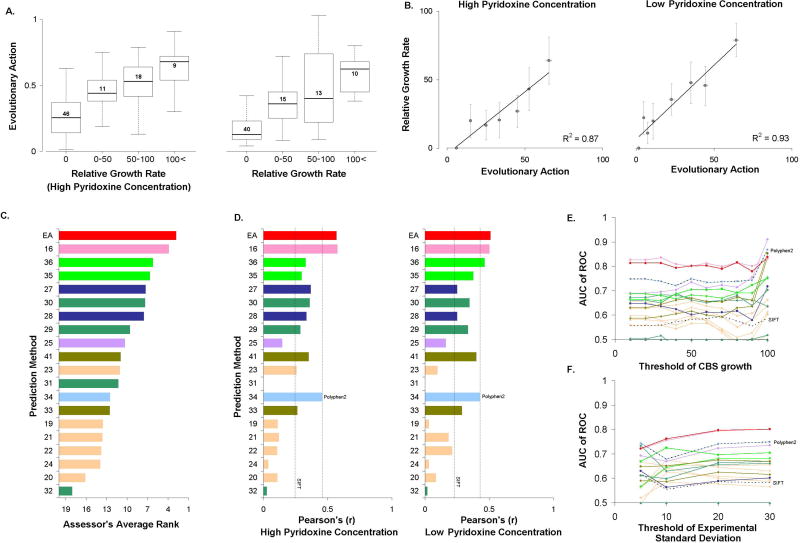
A major challenge in genome interpretation is to estimate the fitness effect of coding variants of unknown significance (VUS). Labor, limited understanding of protein functions, and lack of assays generally limit direct experimental assessment of VUS, and make robust and accurate computational approaches a necessity. Often, however, algorithms that predict mutational effect disagree among themselves and with experimental data, slowing their adoption for clinical diagnostics. To objectively assess such methods, the Critical Assessment of Genome Interpretation (CAGI) community organizes contests to predict unpublished experimental data, available only to CAGI assessors. We review here the CAGI performance of evolutionary action (EA) predictions of mutational impact. EA models the fitness effect of coding mutations analytically, as a product of the gradient of the fitness landscape times the perturbation size. In practice, these terms are computed from phylogenetic considerations as the functional sensitivity of the mutated site and as the magnitude of amino acid substitution, respectively, and yield the percentage loss of wild-type activity. In five CAGI challenges, EA consistently performed on par or better than sophisticated machine learning approaches. This objective assessment suggests that a simple differential model of evolution can interpret the fitness effect of coding variations, opening diverse clinical applications.
Keywords: deleterious and neutral; genetic variation fitness; mutation effect prediction; pathogenic and benign; single-nucleotide polymorphism (SNP); unbiased performance comparison.
© 2017 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA. Epistasis as the primary factor in molecular evolution. Nature. 2012;490(7421):535–538. - PubMed
-
- Buckland M, Gey F. The relationship between recall and precision. Journal of the American society for information science. 1994;45(1):12.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
