

Transcriptomics technologies
- PMID: 28545146
- PMCID: PMC5436640
- DOI: 10.1371/journal.pcbi.1005457
Transcriptomics technologies
Abstract
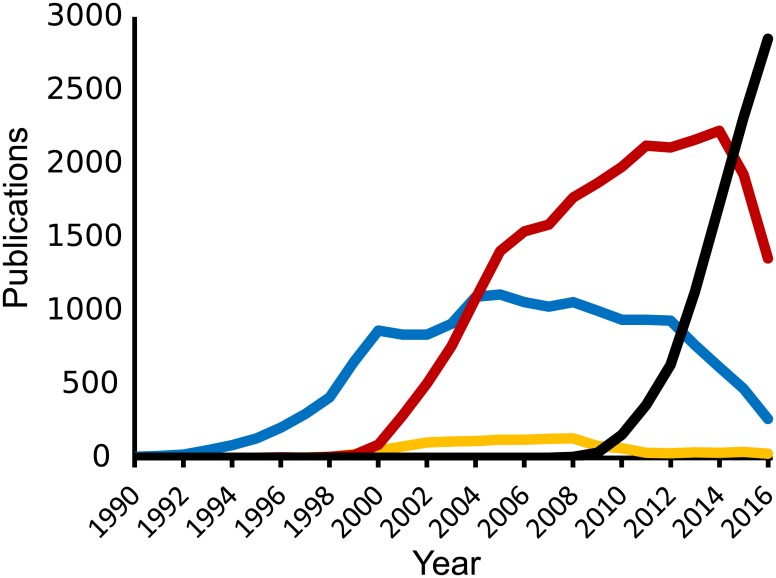
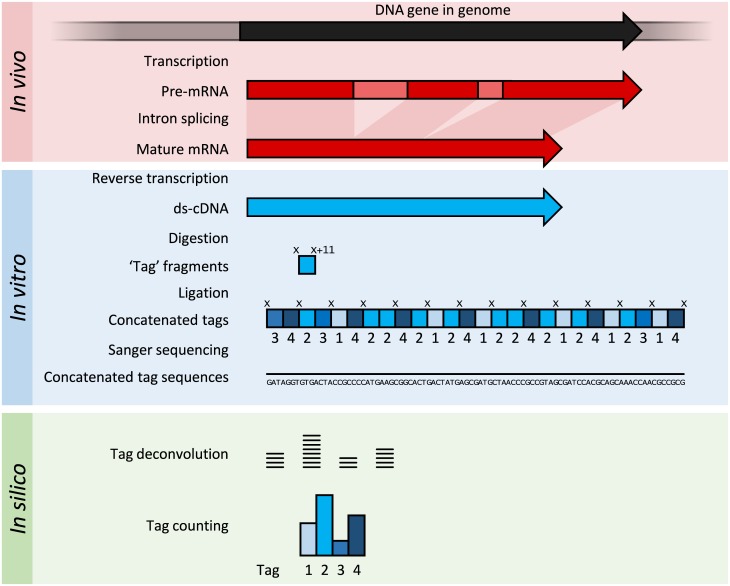
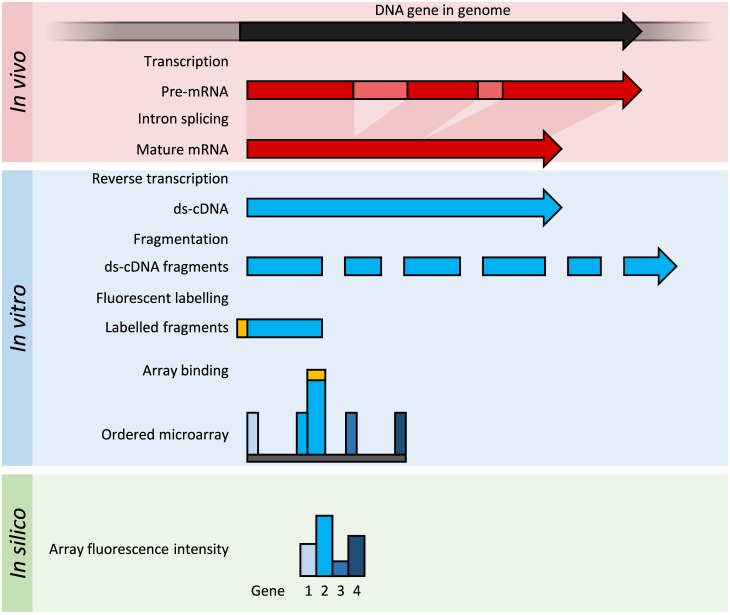
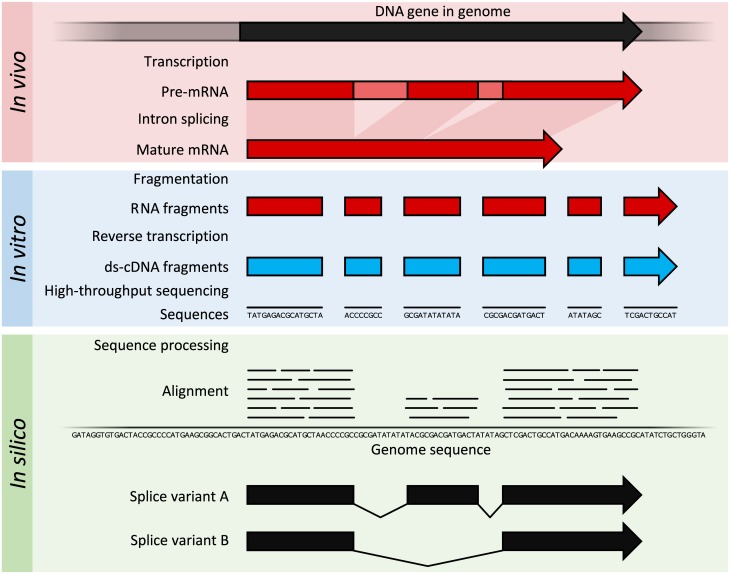
Transcriptomics technologies are the techniques used to study an organism's transcriptome, the sum of all of its RNA transcripts. The information content of an organism is recorded in the DNA of its genome and expressed through transcription. Here, mRNA serves as a transient intermediary molecule in the information network, whilst noncoding RNAs perform additional diverse functions. A transcriptome captures a snapshot in time of the total transcripts present in a cell. The first attempts to study the whole transcriptome began in the early 1990s, and technological advances since the late 1990s have made transcriptomics a widespread discipline. Transcriptomics has been defined by repeated technological innovations that transform the field. There are two key contemporary techniques in the field: microarrays, which quantify a set of predetermined sequences, and RNA sequencing (RNA-Seq), which uses high-throughput sequencing to capture all sequences. Measuring the expression of an organism's genes in different tissues, conditions, or time points gives information on how genes are regulated and reveals details of an organism's biology. It can also help to infer the functions of previously unannotated genes. Transcriptomic analysis has enabled the study of how gene expression changes in different organisms and has been instrumental in the understanding of human disease. An analysis of gene expression in its entirety allows detection of broad coordinated trends which cannot be discerned by more targeted assays.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

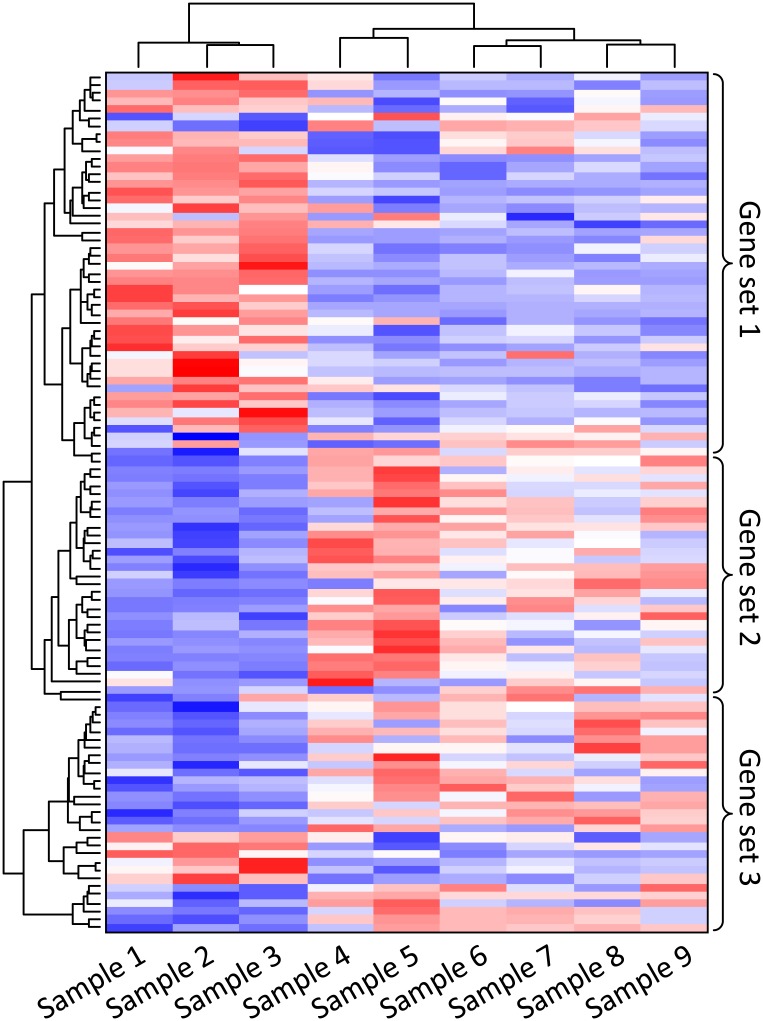
References
-
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, et al. Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991;252:1651–6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
