

Overexpression of Kinase-Dead mTOR Impairs Glucose Homeostasis by Regulating Insulin Secretion and Not β-Cell Mass
- PMID: 28546423
- PMCID: PMC5521866
- DOI: 10.2337/db16-1349
Overexpression of Kinase-Dead mTOR Impairs Glucose Homeostasis by Regulating Insulin Secretion and Not β-Cell Mass
Abstract
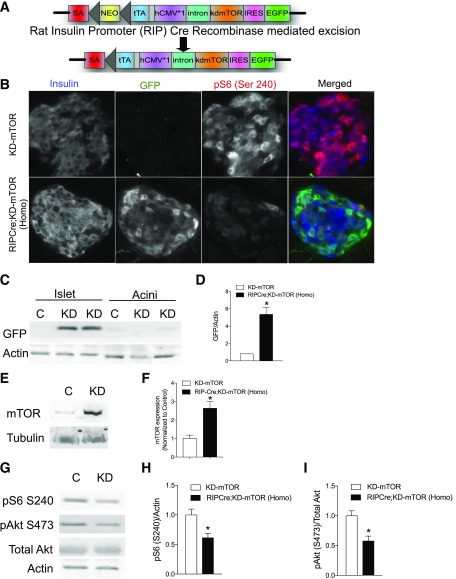
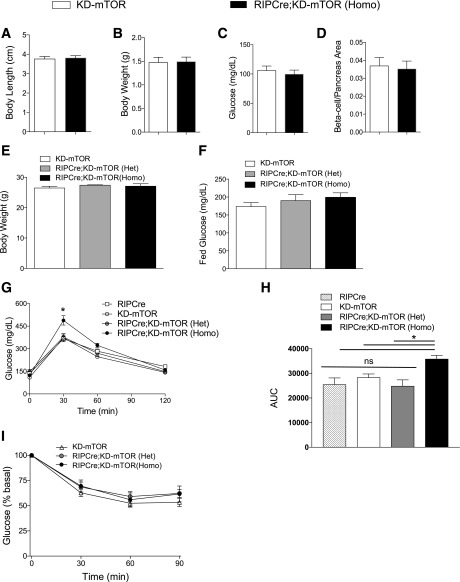
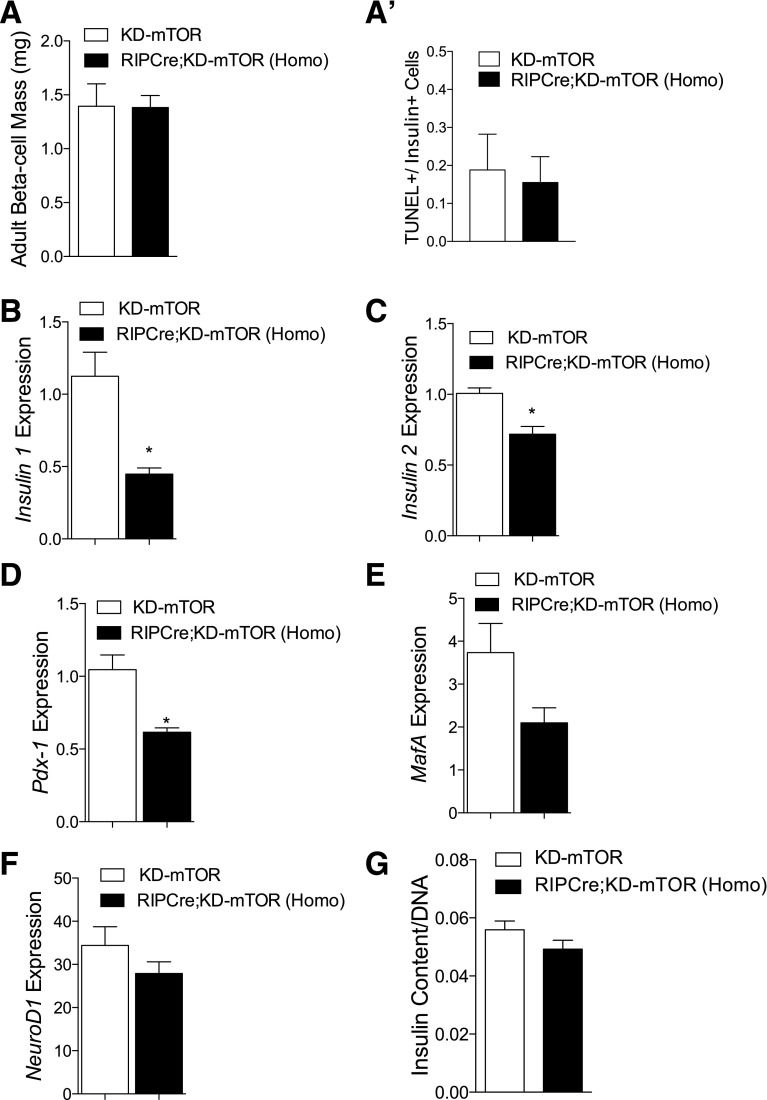
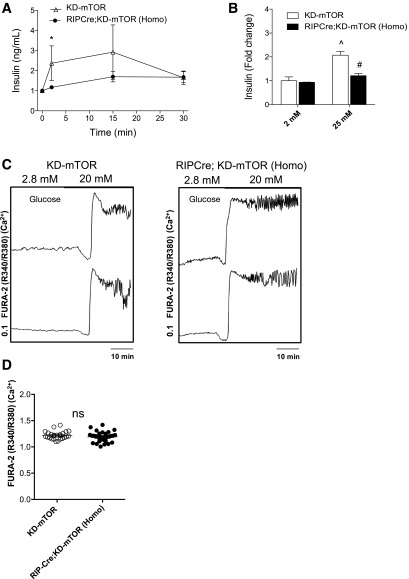
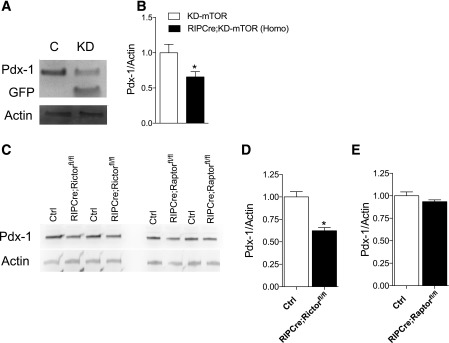
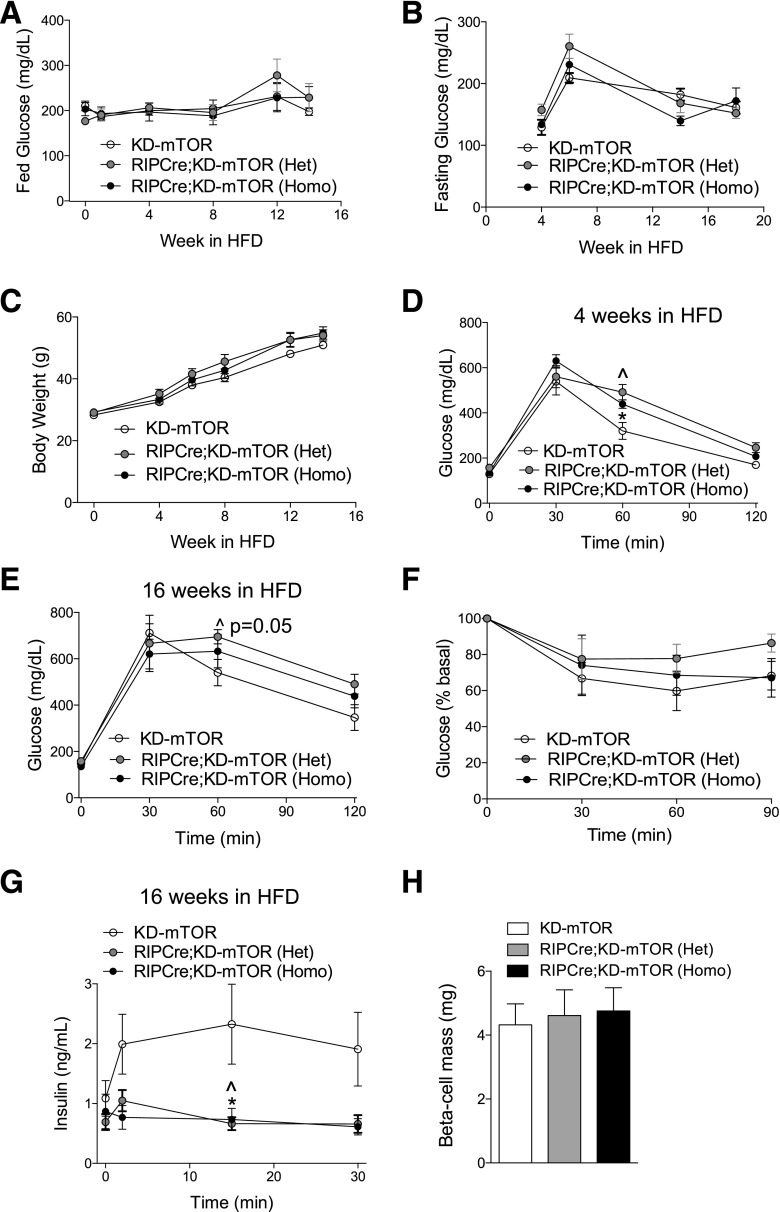
Regulation of glucose homeostasis by insulin depends on β-cell growth and function. Nutrients and growth factor stimuli converge on the conserved protein kinase mechanistic target of rapamycin (mTOR), existing in two complexes, mTORC1 and mTORC2. To understand the functional relevance of mTOR enzymatic activity in β-cell development and glucose homeostasis, we generated mice overexpressing either one or two copies of a kinase-dead mTOR mutant (KD-mTOR) transgene exclusively in β-cells. We examined glucose homeostasis and β-cell function of these mice fed a control chow or high-fat diet. Mice with two copies of the transgene [RIPCre;KD-mTOR (Homozygous)] develop glucose intolerance due to a defect in β-cell function without alterations in β-cell mass with control chow. Islets from RIPCre;KD-mTOR (Homozygous) mice showed reduced mTORC1 and mTORC2 signaling along with transcripts and protein levels of Pdx-1. Islets with reduced mTORC2 signaling in their β-cells (RIPCre;Rictorfl/fl) also showed reduced Pdx-1. When challenged with a high-fat diet, mice carrying one copy of KD-mTOR mutant transgene developed glucose intolerance and β-cell insulin secretion defect but showed no changes in β-cell mass. These findings suggest that the mTOR-mediated signaling pathway is not essential to β-cell growth but is involved in regulating β-cell function in normal and diabetogenic conditions.
© 2017 by the American Diabetes Association.
Figures
References
-
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science 2005;307:1098–1101 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
