

Whole genome sequencing and imputation in isolated populations identify genetic associations with medically-relevant complex traits
- PMID: 28548082
- PMCID: PMC5458552
- DOI: 10.1038/ncomms15606
Whole genome sequencing and imputation in isolated populations identify genetic associations with medically-relevant complex traits
Abstract
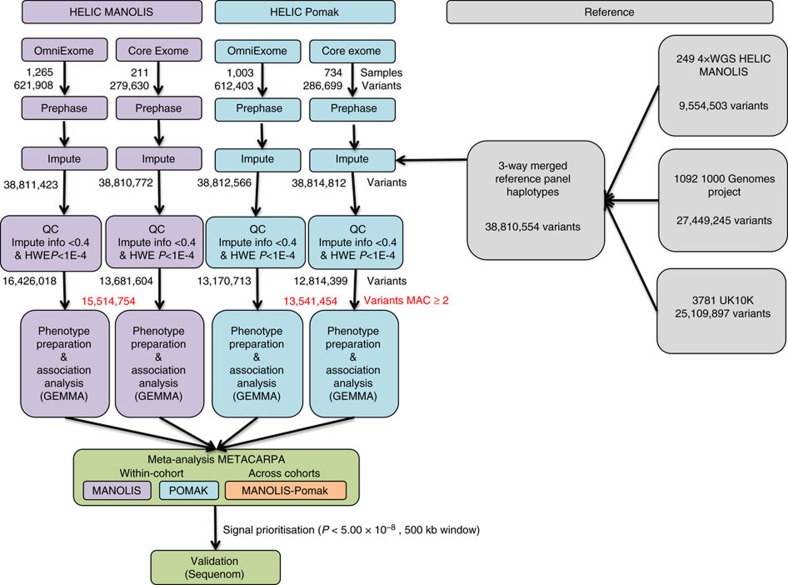
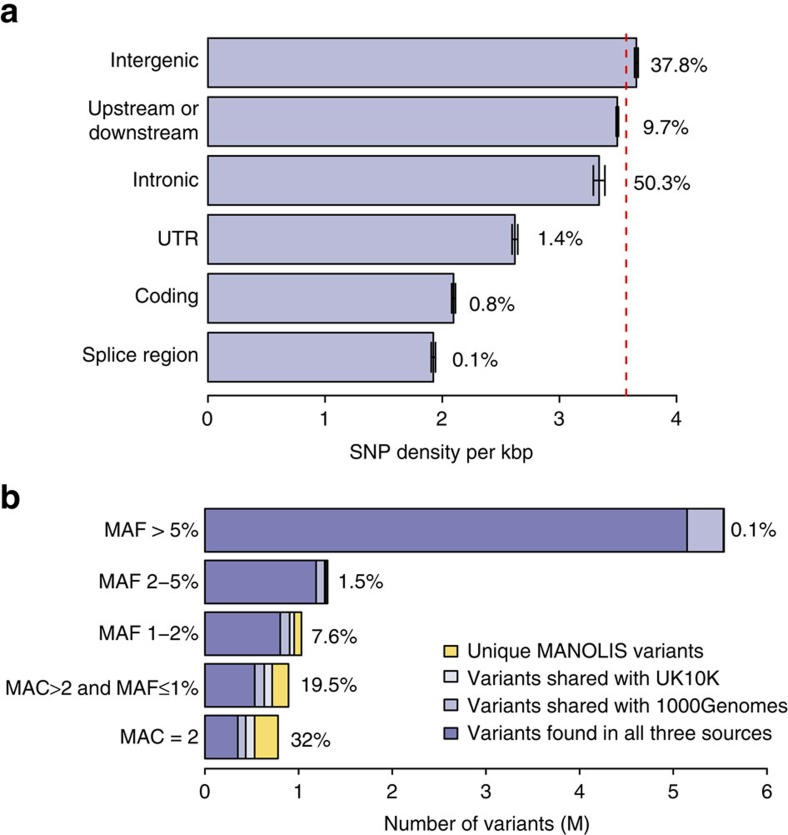
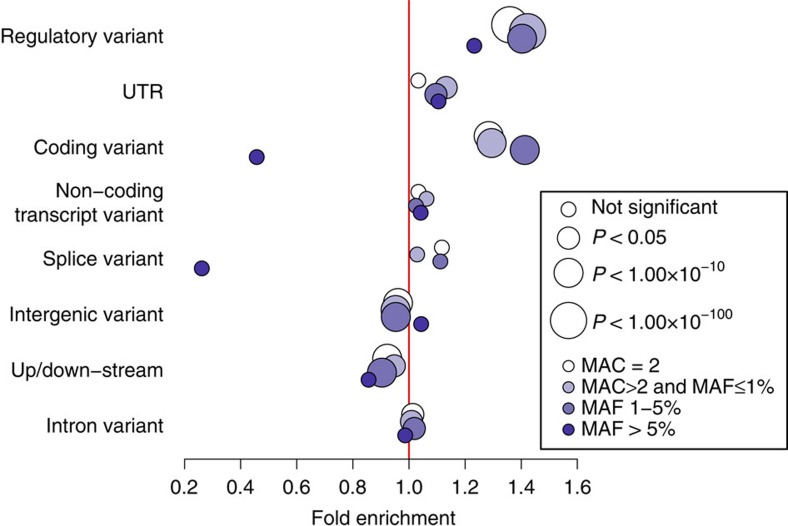
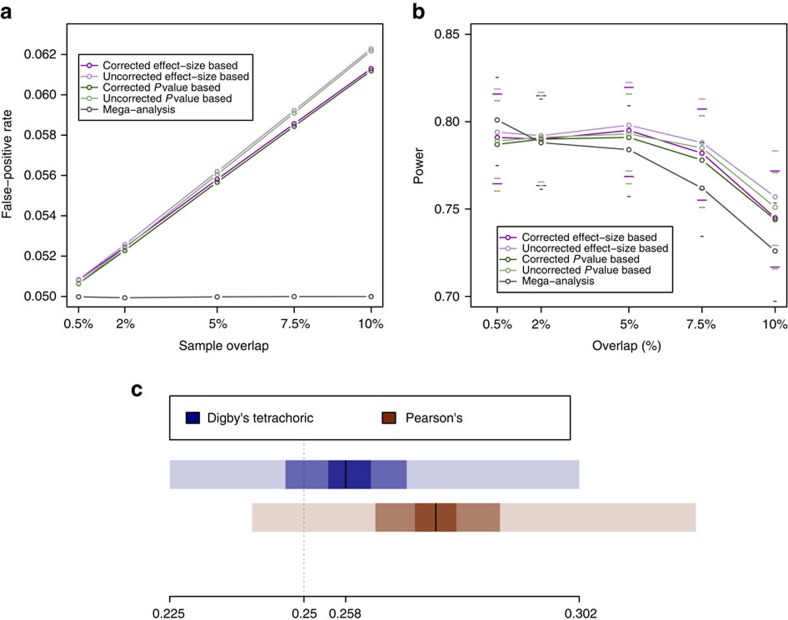
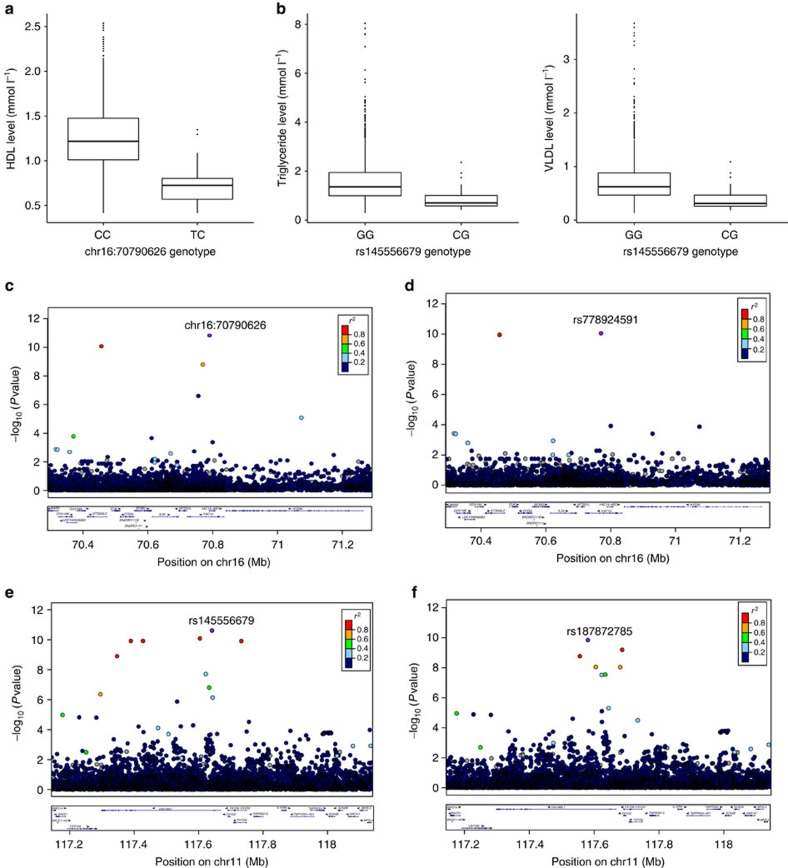
Next-generation association studies can be empowered by sequence-based imputation and by studying founder populations. Here we report ∼9.5 million variants from whole-genome sequencing (WGS) of a Cretan-isolated population, and show enrichment of rare and low-frequency variants with predicted functional consequences. We use a WGS-based imputation approach utilizing 10,422 reference haplotypes to perform genome-wide association analyses and observe 17 genome-wide significant, independent signals, including replicating evidence for association at eight novel low-frequency variant signals. Two novel cardiometabolic associations are at lead variants unique to the founder population sequences: chr16:70790626 (high-density lipoprotein levels beta -1.71 (SE 0.25), P=1.57 × 10-11, effect allele frequency (EAF) 0.006); and rs145556679 (triglycerides levels beta -1.13 (SE 0.17), P=2.53 × 10-11, EAF 0.013). Our findings add empirical support to the contribution of low-frequency variants in complex traits, demonstrate the advantage of including population-specific sequences in imputation panels and exemplify the power gains afforded by population isolates.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel.Eur J Hum Genet. 2017 Jun;25(7):869-876. doi: 10.1038/ejhg.2017.51. Epub 2017 Apr 12. Eur J Hum Genet. 2017. PMID: 28401899 Free PMC article.
-
Rare variant genotype imputation with thousands of study-specific whole-genome sequences: implications for cost-effective study designs.Eur J Hum Genet. 2015 Jul;23(7):975-83. doi: 10.1038/ejhg.2014.216. Epub 2014 Oct 8. Eur J Hum Genet. 2015. PMID: 25293720 Free PMC article.
-
Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations.PLoS Genet. 2019 Dec 23;15(12):e1008500. doi: 10.1371/journal.pgen.1008500. eCollection 2019 Dec. PLoS Genet. 2019. PMID: 31869403 Free PMC article.
-
Unique roles of rare variants in the genetics of complex diseases in humans.J Hum Genet. 2021 Jan;66(1):11-23. doi: 10.1038/s10038-020-00845-2. Epub 2020 Sep 18. J Hum Genet. 2021. PMID: 32948841 Free PMC article. Review.
-
Genotype Imputation in Genome-Wide Association Studies.Curr Protoc Hum Genet. 2019 Jun;102(1):e84. doi: 10.1002/cphg.84. Curr Protoc Hum Genet. 2019. PMID: 31216114 Review.
Cited by
-
Genetic Epidemiology of Complex Phenotypes.Methods Mol Biol. 2021;2249:335-367. doi: 10.1007/978-1-0716-1138-8_19. Methods Mol Biol. 2021. PMID: 33871853 Review.
-
AMPK activation negatively regulates GDAP1, which influences metabolic processes and circadian gene expression in skeletal muscle.Mol Metab. 2018 Oct;16:12-23. doi: 10.1016/j.molmet.2018.07.004. Epub 2018 Jul 25. Mol Metab. 2018. PMID: 30093355 Free PMC article.
-
Whole-genome sequencing analysis of the cardiometabolic proteome.Nat Commun. 2020 Dec 10;11(1):6336. doi: 10.1038/s41467-020-20079-2. Nat Commun. 2020. PMID: 33303764 Free PMC article.
-
Carotid Intima-Media Thickness: Novel Loci, Sex-Specific Effects, and Genetic Correlations With Obesity and Glucometabolic Traits in UK Biobank.Arterioscler Thromb Vasc Biol. 2020 Feb;40(2):446-461. doi: 10.1161/ATVBAHA.119.313226. Epub 2019 Dec 5. Arterioscler Thromb Vasc Biol. 2020. PMID: 31801372 Free PMC article.
-
Large-scale cross-cancer fine-mapping of the 5p15.33 region reveals multiple independent signals.HGG Adv. 2021 Jul 8;2(3):100041. doi: 10.1016/j.xhgg.2021.100041. Epub 2021 Jun 12. HGG Adv. 2021. PMID: 34355204 Free PMC article.
References
-
- Gudbjartsson D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
