

Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations
- PMID: 28548104
- PMCID: PMC5477513
- DOI: 10.1038/ncomms15290
Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations
Abstract
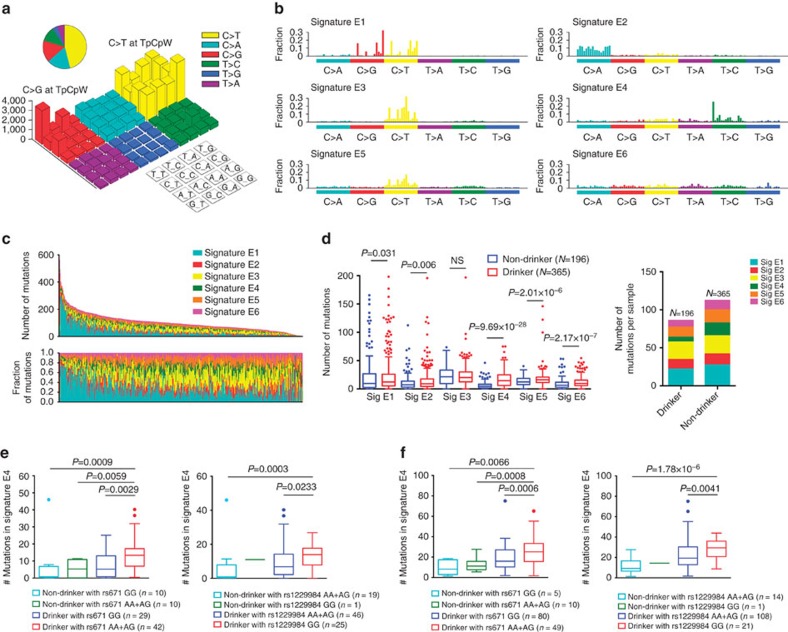
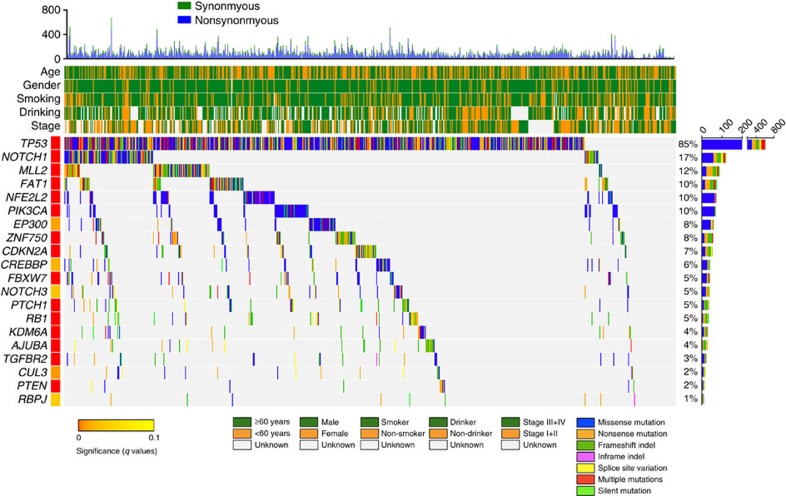
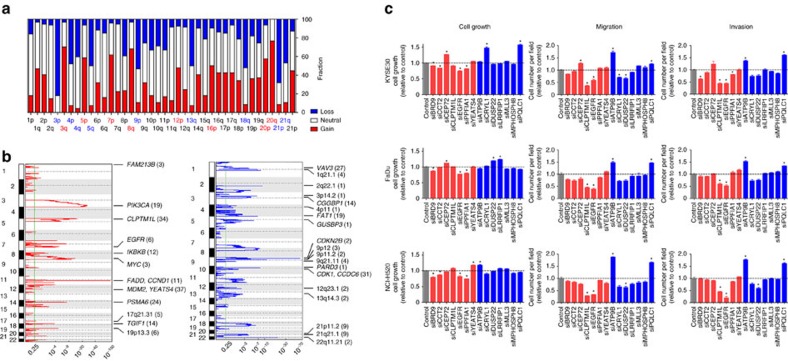
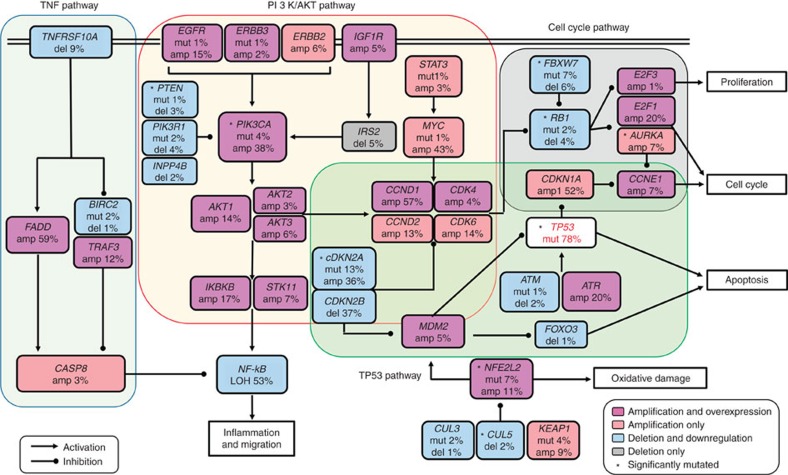
Approximately half of the world's 500,000 new oesophageal squamous-cell carcinoma (ESCC) cases each year occur in China. Here, we show whole-genome sequencing of DNA and RNA in 94 Chinese individuals with ESCC. We identify six mutational signatures (E1-E6), and Signature E4 is unique in ESCC linked to alcohol intake and genetic variants in alcohol-metabolizing enzymes. We discover significantly recurrent mutations in 20 protein-coding genes, 4 long non-coding RNAs and 10 untranslational regions. Functional analyses show six genes that have recurrent copy-number variants in three squamous-cell carcinomas (oesophageal, head and neck and lung) significantly promote cancer cell proliferation, migration and invasion. The most frequently affected genes by structural variation are LRP1B and TTC28. The aberrant cell cycle and PI3K-AKT pathways seem critical in ESCC. These results establish a comprehensive genomic landscape of ESCC and provide potential targets for precision treatment and prevention of the cancer.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

References
-
- Torre L. A. et al. Global cancer statistics, 2012. CA Cancer J. Clin. 65, 87–108 (2015). - PubMed
-
- Chen W. et al. Cancer statistics in China, 2015. CA Cancer J. Clin. 66, 115–132 (2016). - PubMed
-
- Morita M. et al. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: epidemiology, clinical findings, and prevention. Int. J. Clin. Oncol. 15, 126–134 (2010). - PubMed
-
- Toh Y. et al. Alcohol drinking, cigarette smoking, and the development of squamous cell carcinoma of the esophagus: molecular mechanisms of carcinogenesis. Int. J. Clin. Oncol. 15, 135–144 (2010). - PubMed
-
- Islami F. et al. Alcohol drinking and esophageal squamous cell carcinoma with focus on light-drinkers and never-smokers: a systematic review and meta-analysis. Int. J. Cancer 129, 2473–2484 (2011). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
