

Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure
- PMID: 28551825
- PMCID: PMC5589788
- DOI: 10.1007/s00018-017-2547-4
Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure
Abstract
Ferroptosis is a recently recognized caspase-independent form of regulated cell death that is characterized by the accumulation of lethal lipid ROS produced through iron-dependent lipid peroxidation. Considering that regulation of fatty acid metabolism is responsible for the membrane-resident pool of oxidizable fatty acids that undergo lipid peroxidation in ferroptotic processes, we examined the contribution of the key fatty acid metabolism enzyme, acyl-CoA synthetase long-chain family member 4 (ACSL4), in regulating ferroptosis. By using CRISPR/Cas9 technology, we found that knockout of Acsl4 in ferroptosis-sensitive murine and human cells conferred protection from erastin- and RSL3-induced cell death. In the same cell types, deletion of mixed lineage kinase domain-like (Mlkl) blocked susceptibility to necroptosis, as expected. Surprisingly, these studies also revealed ferroptosis and necroptosis are alternative, in that resistance to one pathway sensitized cells to death via the other pathway. These data suggest a mechanism by which one regulated necrosis pathway compensates for another when either ferroptosis or necroptosis is compromised. We verified the synergistic contributions of ferroptosis and necroptosis to tissue damage during acute organ failure in vivo. Interestingly, in the course of pathophysiological acute ischemic kidney injury, ACSL4 was initially upregulated and its expression level correlated with the severity of tissue damage. Together, our findings reveal ACSL4 to be a reliable biomarker of the emerging cell death modality of ferroptosis, which may also serve as a novel therapeutic target in preventing pathological cell death processes.
Keywords: ACSL4; Ferroptosis; Ischemia-reperfusion injury; MLKL; Necroptosis.
Conflict of interest statement
The authors declare no competing or financial interests.
Figures
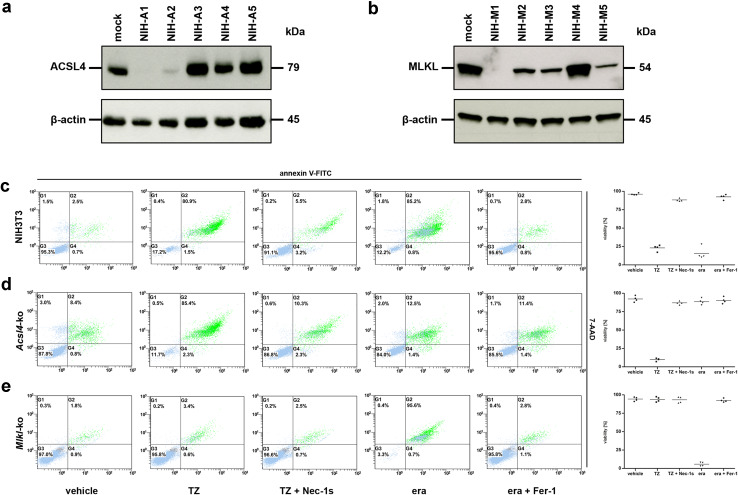
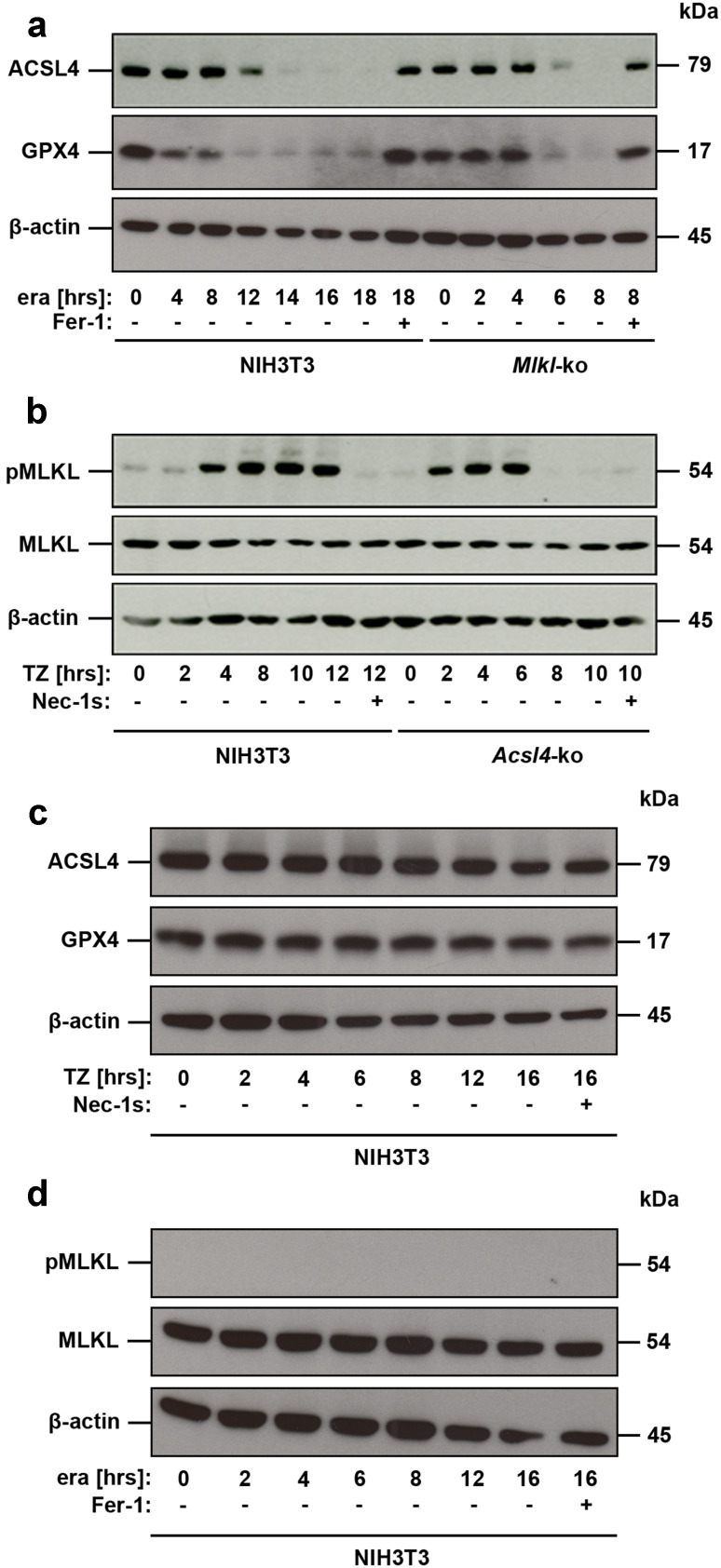
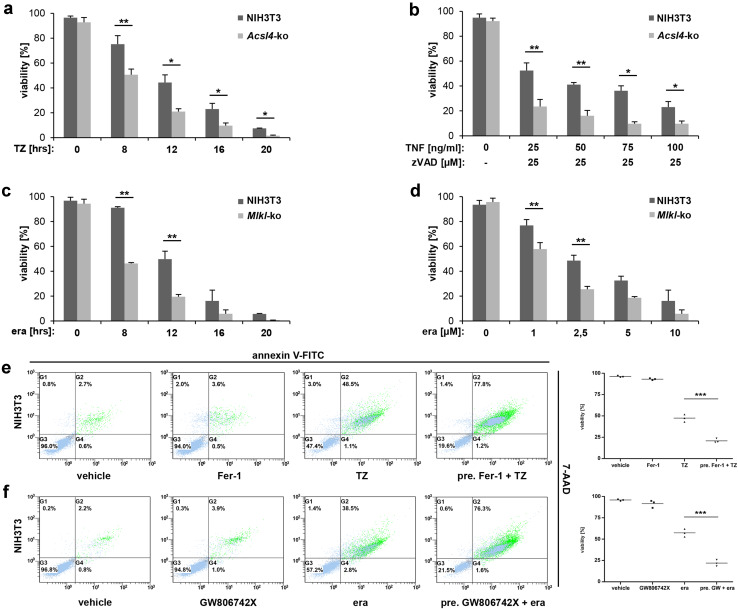


References
Publication types
MeSH terms
Substances
Grants and funding
- 1057905/National Health and Medical Research Council/International
- R35 CA209896/CA/NCI NIH HHS/United States
- 1067289/National Health and Medical Research Council/International
- IRII9000220/National Health and Medical Research Council/International
- R01CA09706/National Institutes of Health (US)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
