

Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics
- PMID: 28552170
- PMCID: PMC5448308
- DOI: 10.1016/j.pnmrs.2017.01.001
Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics
Abstract
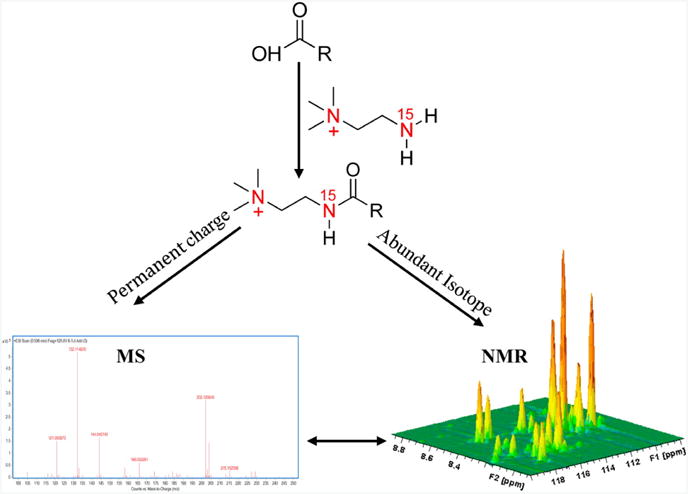
Metabolomics is undergoing tremendous growth and is being employed to solve a diversity of biological problems from environmental issues to the identification of biomarkers for human diseases. Nuclear magnetic resonance (NMR) and mass spectrometry (MS) are the analytical tools that are routinely, but separately, used to obtain metabolomics data sets due to their versatility, accessibility, and unique strengths. NMR requires minimal sample handling without the need for chromatography, is easily quantitative, and provides multiple means of metabolite identification, but is limited to detecting the most abundant metabolites (⩾1μM). Conversely, mass spectrometry has the ability to measure metabolites at very low concentrations (femtomolar to attomolar) and has a higher resolution (∼103-104) and dynamic range (∼103-104), but quantitation is a challenge and sample complexity may limit metabolite detection because of ion suppression. Consequently, liquid chromatography (LC) or gas chromatography (GC) is commonly employed in conjunction with MS, but this may lead to other sources of error. As a result, NMR and mass spectrometry are highly complementary, and combining the two techniques is likely to improve the overall quality of a study and enhance the coverage of the metabolome. While the majority of metabolomic studies use a single analytical source, there is a growing appreciation of the inherent value of combining NMR and MS for metabolomics. An overview of the current state of utilizing both NMR and MS for metabolomics will be presented.
Copyright © 2017 Elsevier B.V. All rights reserved.
Figures

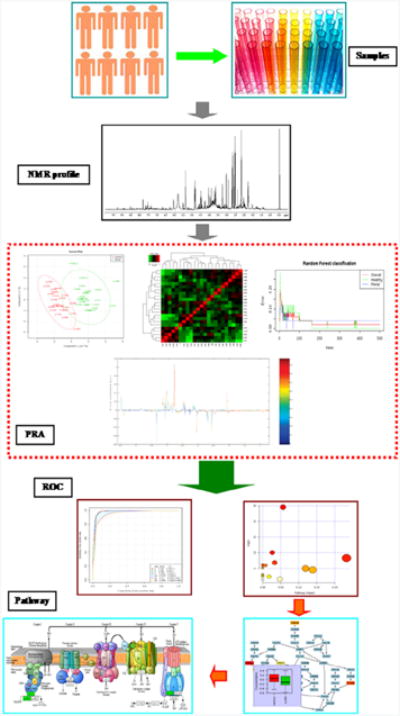





 ), Rotenone (
), Rotenone (
 ), 6-OHDA (
), 6-OHDA (
 ), MPP+ (
), MPP+ (
 ), and Paraquat (
), and Paraquat (
 ). Corresponding dendrograms are shown in (D-F). The statistical significance of each node in the dendrogram is indicated by a p value. Reproduced with permission from reference [57].
). Corresponding dendrograms are shown in (D-F). The statistical significance of each node in the dendrogram is indicated by a p value. Reproduced with permission from reference [57].References
-
- Wachtel-Galor S, Benzie IFF. Herbal Medicine: An Introduction to Its History, Usage, Regulation, Current Trends, and Research Needs. 2011 - PubMed
-
- Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998;16:373–378. - PubMed
-
- Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29:1181–1189. - PubMed
-
- Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst. 2005;130:606–625. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
