

Autophagy as an Emerging Common Pathomechanism in Inherited Peripheral Neuropathies
- PMID: 28553203
- PMCID: PMC5425483
- DOI: 10.3389/fnmol.2017.00143
Autophagy as an Emerging Common Pathomechanism in Inherited Peripheral Neuropathies
Abstract
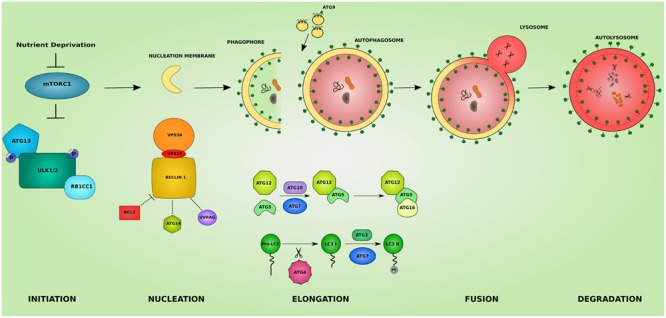
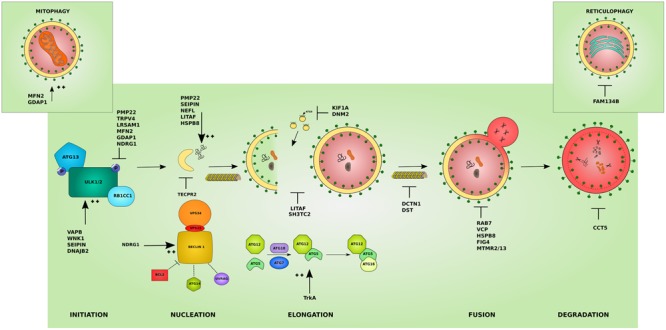
The inherited peripheral neuropathies (IPNs) comprise a growing list of genetically heterogeneous diseases. With mutations in more than 80 genes being reported to cause IPNs, a wide spectrum of functional consequences is expected to follow this genotypic diversity. Hence, the search for a common pathomechanism among the different phenotypes has become the holy grail of functional research into IPNs. During the last decade, studies on several affected genes have shown a direct and/or indirect correlation with autophagy. Autophagy, a cellular homeostatic process, is required for the removal of cell aggregates, long-lived proteins and dead organelles from the cell in double-membraned vesicles destined for the lysosomes. As an evolutionarily highly conserved process, autophagy is essential for the survival and proper functioning of the cell. Recently, neuronal cells have been shown to be particularly vulnerable to disruption of the autophagic pathway. Furthermore, autophagy has been shown to be affected in various common neurodegenerative diseases of both the central and the peripheral nervous system including Alzheimer's, Parkinson's, and Huntington's diseases. In this review we provide an overview of the genes involved in hereditary neuropathies which are linked to autophagy and we propose the disruption of the autophagic flux as an emerging common pathomechanism. We also shed light on the different steps of the autophagy pathway linked to these genes. Finally, we review the concept of autophagy being a therapeutic target in IPNs, and the possibilities and challenges of this pathway-specific targeting.
Keywords: Charcot-Marie-Tooth; autophagy; hereditary neuropathies; neurodegeneration; proteostasis.
Figures
References
-
- Axe E. L., Walker S. A., Manifava M., Chandra P., Roderick H. L., Habermann A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182 685–701. 10.1083/jcb.200803137 - DOI - PMC - PubMed
-
- Azzedine H., Bolino A., Taïeb T., Birouk N., Di Duca M., Bouhouche A., et al. (2003). Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am. J. Hum. Genet. 72 1141–1153. 10.1086/375034 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials