

A Review on Recent Computational Methods for Predicting Noncoding RNAs
- PMID: 28553651
- PMCID: PMC5434267
- DOI: 10.1155/2017/9139504
A Review on Recent Computational Methods for Predicting Noncoding RNAs
Abstract
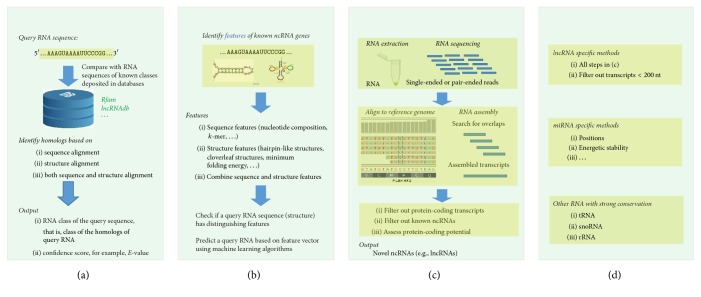
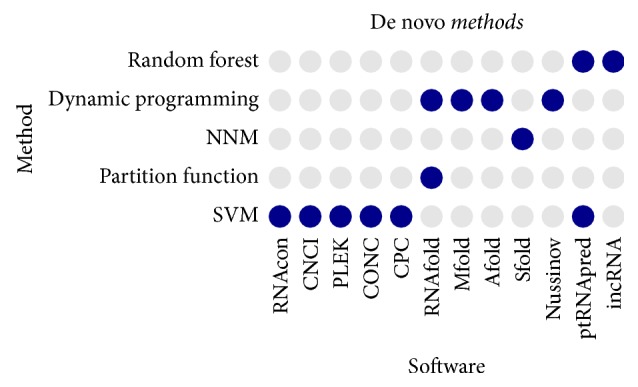
Noncoding RNAs (ncRNAs) play important roles in various cellular activities and diseases. In this paper, we presented a comprehensive review on computational methods for ncRNA prediction, which are generally grouped into four categories: (1) homology-based methods, that is, comparative methods involving evolutionarily conserved RNA sequences and structures, (2) de novo methods using RNA sequence and structure features, (3) transcriptional sequencing and assembling based methods, that is, methods designed for single and pair-ended reads generated from next-generation RNA sequencing, and (4) RNA family specific methods, for example, methods specific for microRNAs and long noncoding RNAs. In the end, we summarized the advantages and limitations of these methods and pointed out a few possible future directions for ncRNA prediction. In conclusion, many computational methods have been demonstrated to be effective in predicting ncRNAs for further experimental validation. They are critical in reducing the huge number of potential ncRNAs and pointing the community to high confidence candidates. In the future, high efficient mapping technology and more intrinsic sequence features (e.g., motif and k-mer frequencies) and structure features (e.g., minimum free energy, conserved stem-loop, or graph structures) are suggested to be combined with the next- and third-generation sequencing platforms to improve ncRNA prediction.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
