

Oxidative pathways in the sickle cell and beyond
- PMID: 28554826
- PMCID: PMC5696113
- DOI: 10.1016/j.bcmd.2017.05.009
Oxidative pathways in the sickle cell and beyond
Abstract
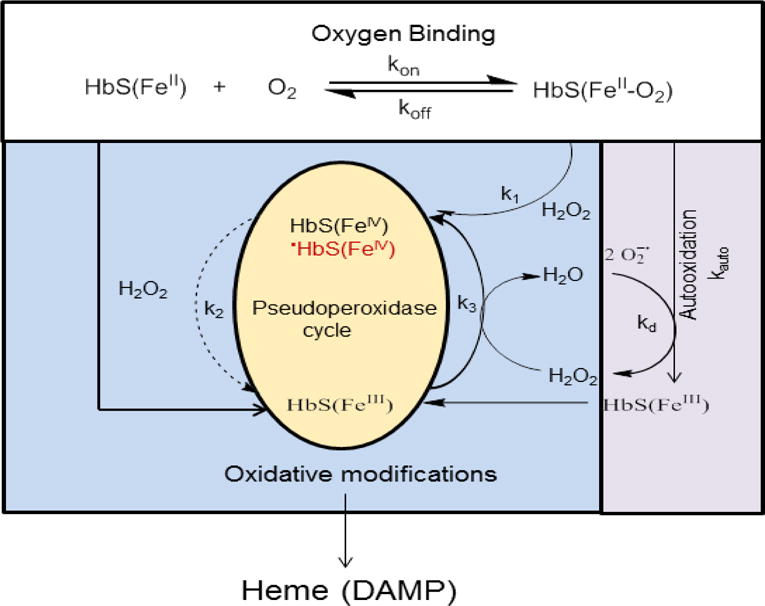
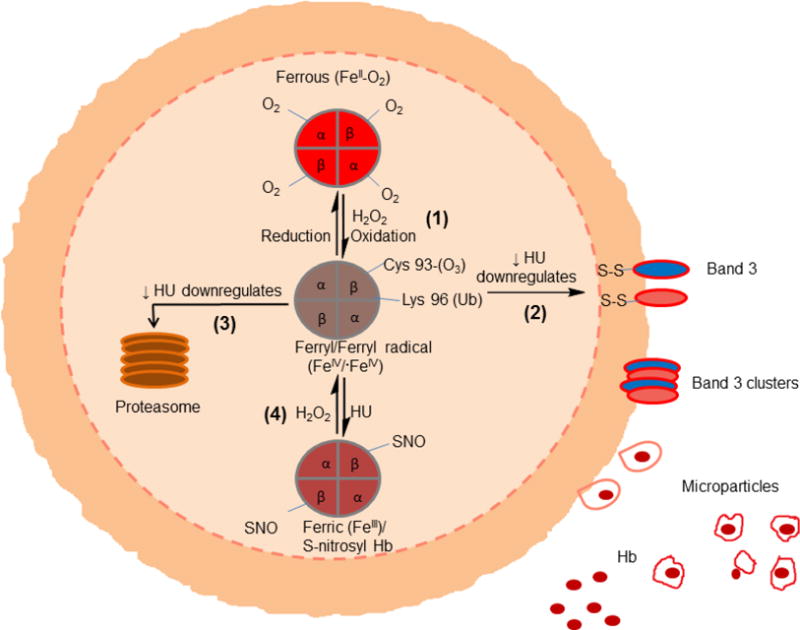
Polymerization of deoxy sickle cell hemoglobin (HbS) is well recognized as the primary event that triggers the classic cycles of sickling/unsickling of patients red blood cells (RBCs). RBCs are also subjected to continuous endogenous and exogenous oxidative onslaughts resulting in hemolytic rate increases which contribute to the evolution of vasculopathies associated with this disease. Compared to steady-state conditions, the occurrences of vaso-occlusive crises increase the levels of both RBC-derived microparticles as well as extracellular Hb in circulation. Common byproduct resulting from free Hb oxidation and from Hb-laden microparticles is heme (now recognized as damage associated molecular pattern (DAMP) molecule) which has been shown to initiate inflammatory responses. This review provides new insights into the interplay between microparticles, free Hb and heme focusing on Hb's pseudoperoxidative activity that drives RBC's cytosolic, membrane changes as well as oxidative toxicity towards the vascular system. Emerging antioxidative strategies that include the use of protein and heme scavengers in controlling Hb oxidative pathways are discussed.
Keywords: Ferryl hemoglobin; Heme oxidation; Microparticles; Pseudoperoxidase; Sickle cell hemoglobin.
Published by Elsevier Inc.
Figures
References
-
- Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science. 1949;110:543–548. - PubMed
-
- Dickerson RE, Gies I. Hemoglobin: structure, function, evolution, and pathology, The Benjamin/Cummings Publishing Company, California. 1983
-
- Hebbel RP. Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin North Am. 2014;28:181–198. - PubMed
-
- Wood KC, Granger DN. sickle cell disease: role for reactive oxygen and nitrogen metabolites. Clin Exp Pharm Physiol. 2007;34:926–932. - PubMed
-
- Connes PI, Lamarre Y, Waltz X, Ballas SK, Lemonne N, et al. Haemolysis and abnormal haemorheology in sickle cell anaemia. Br J Haematol. 2014;165:564–572. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
