

Genomic determinants of chronic myelomonocytic leukemia
- PMID: 28555081
- PMCID: PMC8370480
- DOI: 10.1038/leu.2017.164
Genomic determinants of chronic myelomonocytic leukemia
Abstract
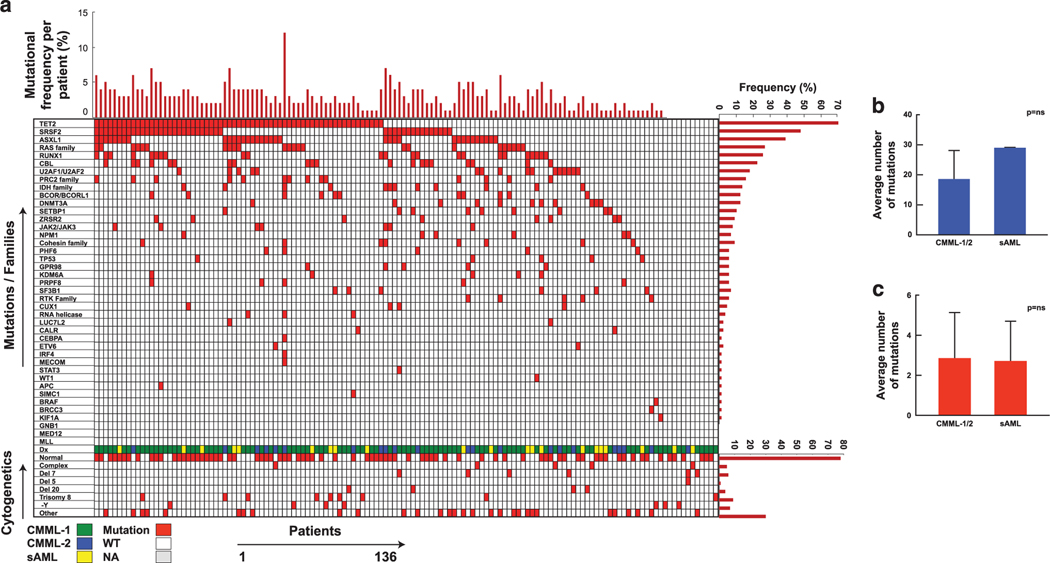
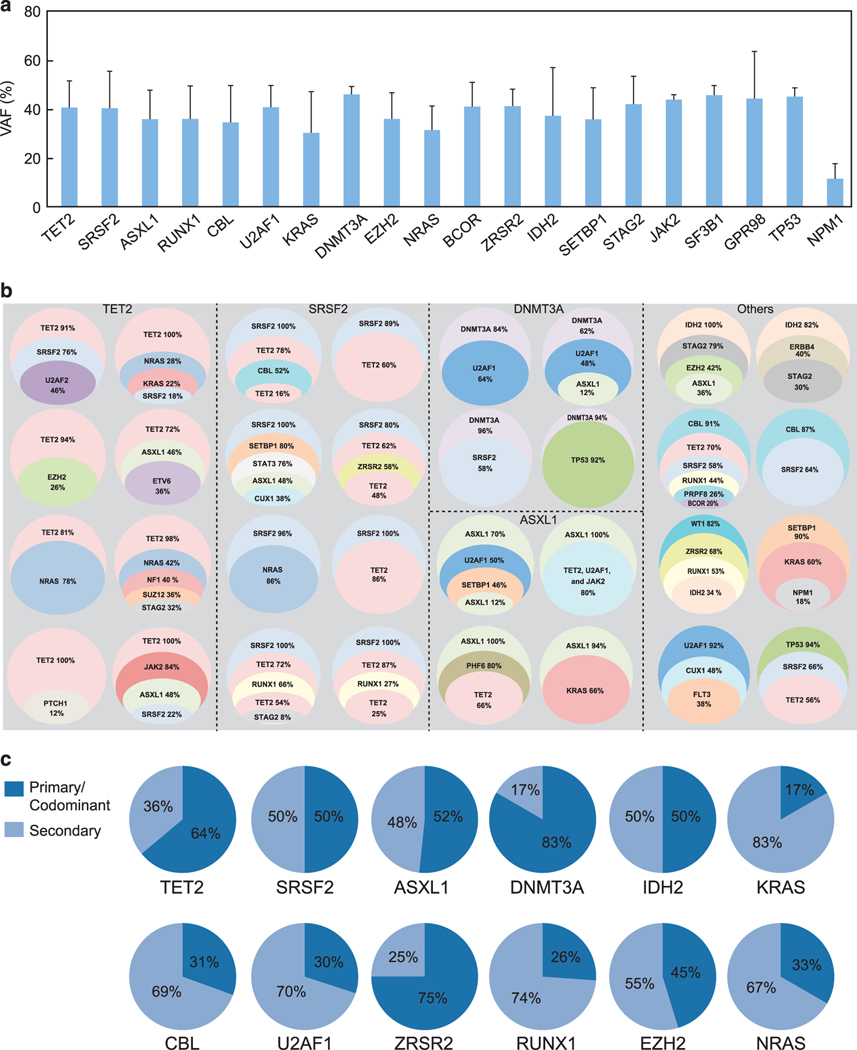
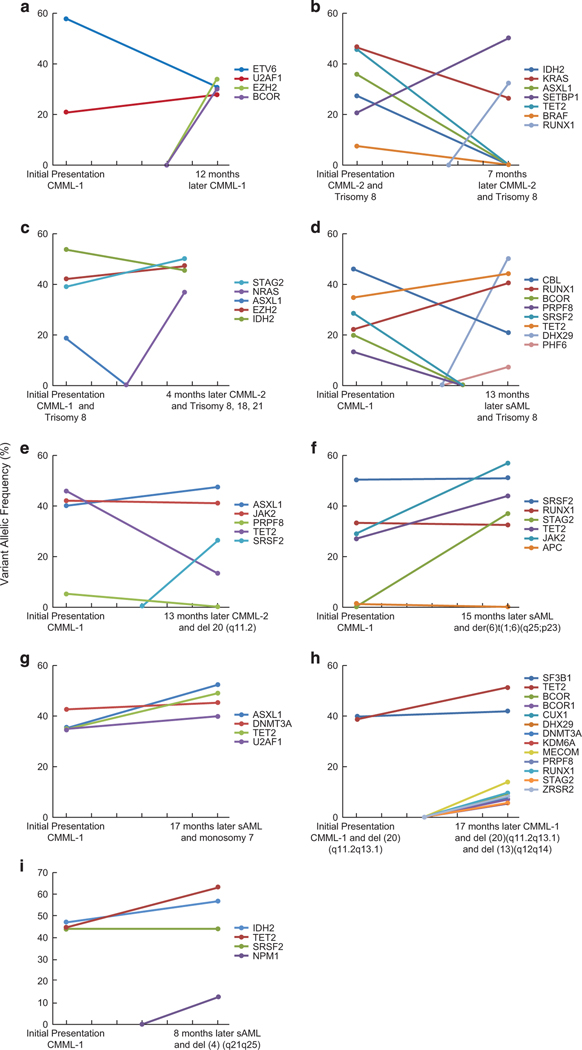
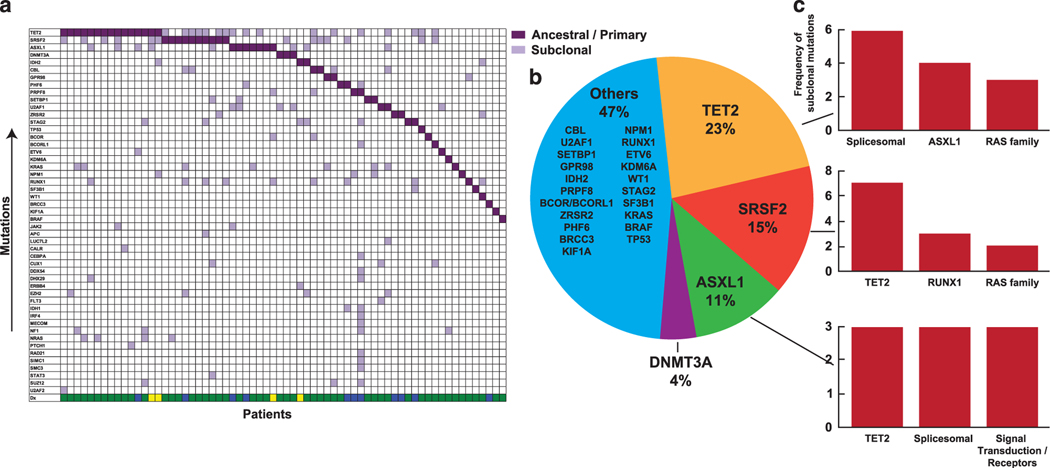
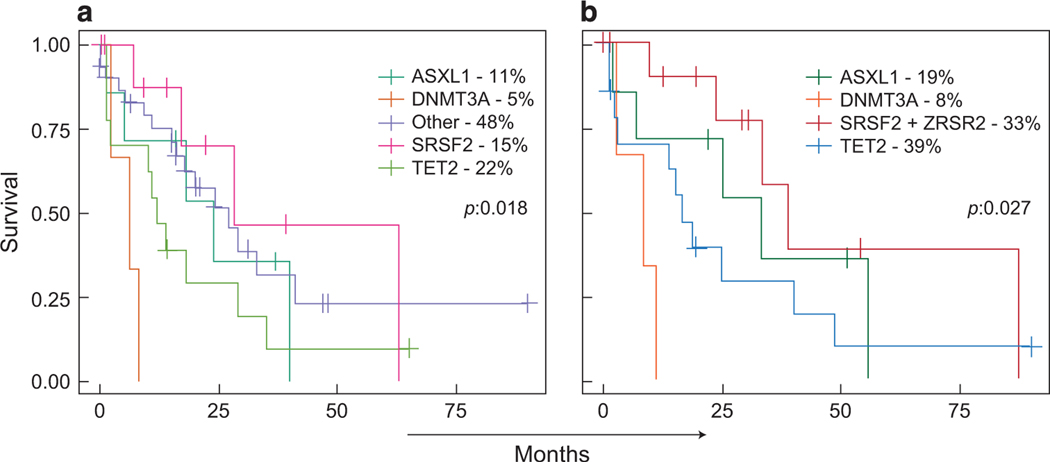
The biology, clinical phenotype and progression rate of chronic myelomonocytic leukemia (CMML) are highly variable due to diverse initiating and secondary clonal genetic events. To determine the effects of molecular features including clonal hierarchy in CMML, we studied whole-exome and targeted next-generation sequencing data from 150 patients with robust clinical and molecular annotation assessed cross-sectionally and at serial time points of disease evolution. To identify molecular lesions unique to CMML, we compared it to the related myeloid neoplasms (N=586), including juvenile myelomonocytic leukemia, myelodysplastic syndromes (MDS) and primary monocytic acute myeloid leukemia and discerned distinct molecular profiles despite similar pathomorphological features. Within CMML, mutations in certain pathways correlated with clinical classification, for example, proliferative vs dysplastic features. While most CMML patients (59%) had ancestral (dominant/co-dominant) mutations involving TET2, SRSF2 or ASXL1 genes, secondary subclonal hierarchy correlated with clinical phenotypes or outcomes. For example, progression was associated with acquisition of new expanding clones carrying biallelic TET2 mutations or RAS family, or spliceosomal gene mutations. In contrast, dysplastic features correlated with mutations usually encountered in MDS (for example, SF3B1 and U2AF1). Classification of CMML based on hierarchies of ancestral and subclonal mutational events may correlate strongly with clinical features and prognosis.
Conflict of interest statement
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Figures

References
-
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391–2405. - PubMed
-
- Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M et al.Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 2013; 45: 937–941. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
