

Rapid amyloid-β oligomer and protofibril accumulation in traumatic brain injury
- PMID: 28557010
- PMCID: PMC8028271
- DOI: 10.1111/bpa.12532
Rapid amyloid-β oligomer and protofibril accumulation in traumatic brain injury
Abstract
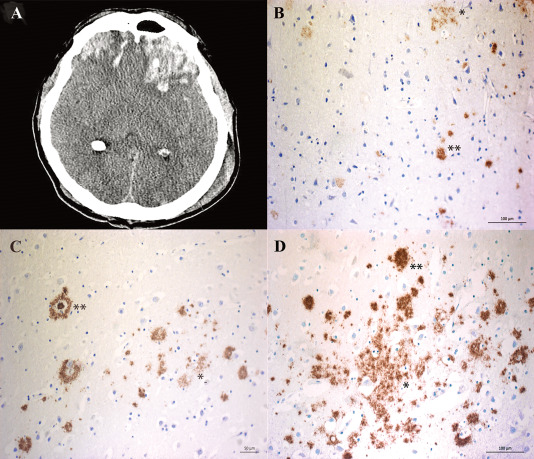
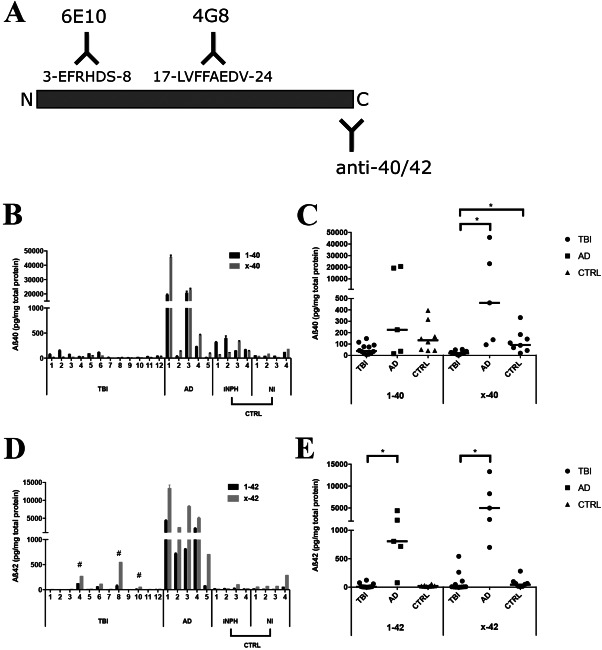
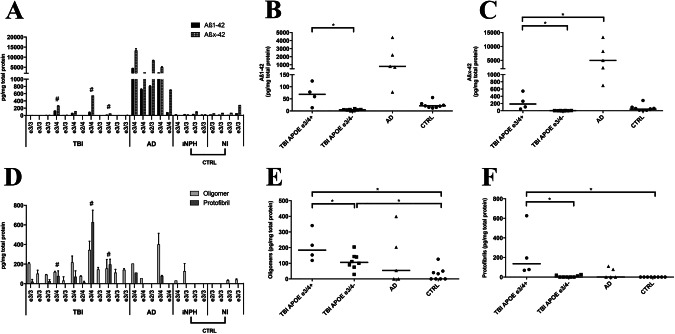
Deposition of amyloid-β (Aβ) is central to Alzheimer's disease (AD) pathogenesis and associated with progressive neurodegeneration in traumatic brain injury (TBI). We analyzed predisposing factors for Aβ deposition including monomeric Aβ40, Aβ42 and Aβ oligomers/protofibrils, Aβ species with pronounced neurotoxic properties, following human TBI. Highly selective ELISAs were used to analyze N-terminally intact and truncated Aβ40 and Aβ42, as well as Aβ oligomers/protofibrils, in human brain tissue, surgically resected from severe TBI patients (n = 12; mean age 49.5 ± 19 years) due to life-threatening brain swelling/hemorrhage within one week post-injury. The TBI tissues were compared to post-mortem AD brains (n = 5), to post-mortem tissue of neurologically intact (NI) subjects (n = 4) and to cortical biopsies obtained at surgery for idiopathic normal pressure hydrocephalus patients (iNPH; n = 4). The levels of Aβ40 and Aβ42 were not elevated by TBI. The levels of Aβ oligomers/protofibrils in TBI were similar to those in the significantly older AD patients and increased compared to NI and iNPH controls (P < 0.05). Moreover, TBI patients carrying the AD risk genotype Apolipoprotein E epsilon3/4 (APOE ε3/4; n = 4) had increased levels of Aβ oligomers/protofibrils (P < 0.05) and of both N-terminally intact and truncated Aβ42 (P < 0.05) compared to APOE ε3/4-negative TBI patients (n = 8). Neuropathological analysis showed insoluble Aβ aggregates (commonly referred to as Aβ plaques) in three TBI patients, all of whom were APOE ε3/4 carriers. We conclude that soluble intermediary Aβ aggregates form rapidly after TBI, especially among APOE ε3/4 carriers. Further research is needed to determine whether these aggregates aggravate the clinical short- and long-term outcome in TBI.
Keywords: Alzheimer's disease; amyloid β oligomers; amyloid-β; traumatic brain injury.
© 2017 International Society of Neuropathology.
Conflict of interest statement
ERW and LS are employees of BioArctic AB. CM and HB are employees of BioArctic AB and own shares in the company. LL is a co‐founder and chairman of the board of BioArctic AB and owns shares in the company. The remaining authors declare no competing financial interests.
Figures

Comment in
-
Traumatic brain injury: a platform for studies in Aβ processing: Commentary on: "Rapid Aβ oligomer and protofibril accumulation in traumatic brain injury".Brain Pathol. 2018 Jul;28(4):463-465. doi: 10.1111/bpa.12534. Brain Pathol. 2018. PMID: 30133864 Free PMC article. No abstract available.
References
-
- Benilova I, Karran E, De Strooper B (2012) The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci 15:349–357. - PubMed
-
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. - PubMed
-
- Casoli T, Di Stefano G, Giorgetti B, Grossi Y, Balietti M, Fattoretti P, Bertoni‐Freddari C (2007) Release of beta‐amyloid from high‐density platelets: implications for Alzheimer's disease pathology. Ann New York Acad Sci 1096:170–178. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
