

X-ray Scattering Studies of Protein Structural Dynamics
- PMID: 28558231
- PMCID: PMC5562295
- DOI: 10.1021/acs.chemrev.6b00790
X-ray Scattering Studies of Protein Structural Dynamics
Abstract
X-ray scattering is uniquely suited to the study of disordered systems and thus has the potential to provide insight into dynamic processes where diffraction methods fail. In particular, while X-ray crystallography has been a staple of structural biology for more than half a century and will continue to remain so, a major limitation of this technique has been the lack of dynamic information. Solution X-ray scattering has become an invaluable tool in structural and mechanistic studies of biological macromolecules where large conformational changes are involved. Such systems include allosteric enzymes that play key roles in directing metabolic fluxes of biochemical pathways, as well as large, assembly-line type enzymes that synthesize secondary metabolites with pharmaceutical applications. Furthermore, crystallography has the potential to provide information on protein dynamics via the diffuse scattering patterns that are overlaid with Bragg diffraction. Historically, these patterns have been very difficult to interpret, but recent advances in X-ray detection have led to a renewed interest in diffuse scattering analysis as a way to probe correlated motions. Here, we will review X-ray scattering theory and highlight recent advances in scattering-based investigations of protein solutions and crystals, with a particular focus on complex enzymes.
Figures






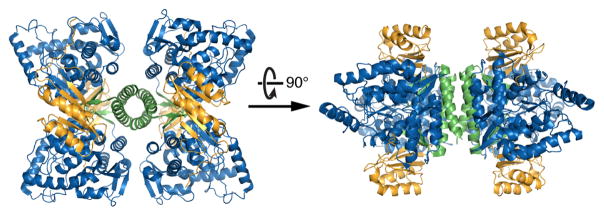













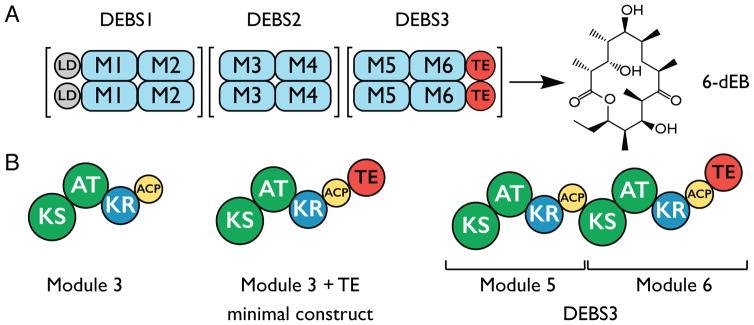





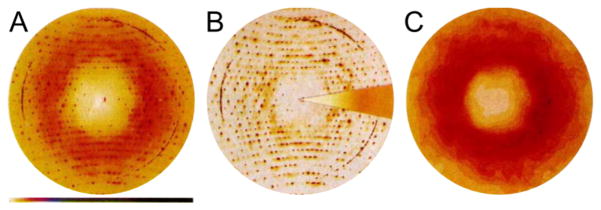






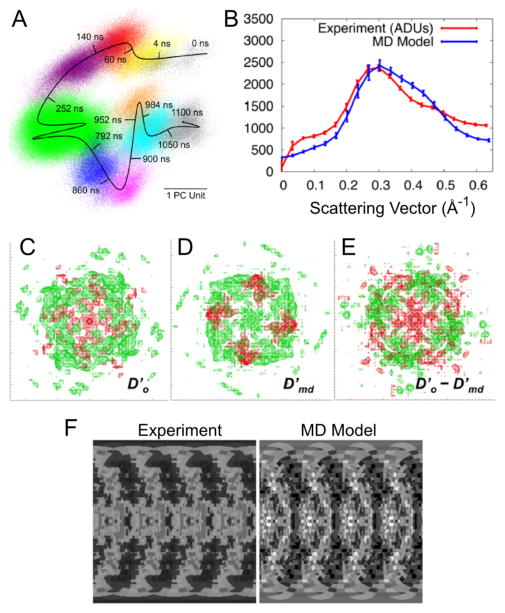








References
-
- Svergun D, Barberato C, Koch MHJ. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773.
-
- Caspar DLD, Clarage J, Salunke DM, Clarage M. Liquid-like movements in crystalline insulin. Nature. 1988;332:659–662. - PubMed
-
- Moore PB. On the relationship between diffraction patterns and motions in macro-molecular crystals. Structure. 2009;17:1307–1315. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
