

C-X-C Motif Chemokine Receptor 3 Splice Variants Differentially Activate Beta-Arrestins to Regulate Downstream Signaling Pathways
- PMID: 28559424
- PMCID: PMC5508197
- DOI: 10.1124/mol.117.108522
C-X-C Motif Chemokine Receptor 3 Splice Variants Differentially Activate Beta-Arrestins to Regulate Downstream Signaling Pathways
Abstract
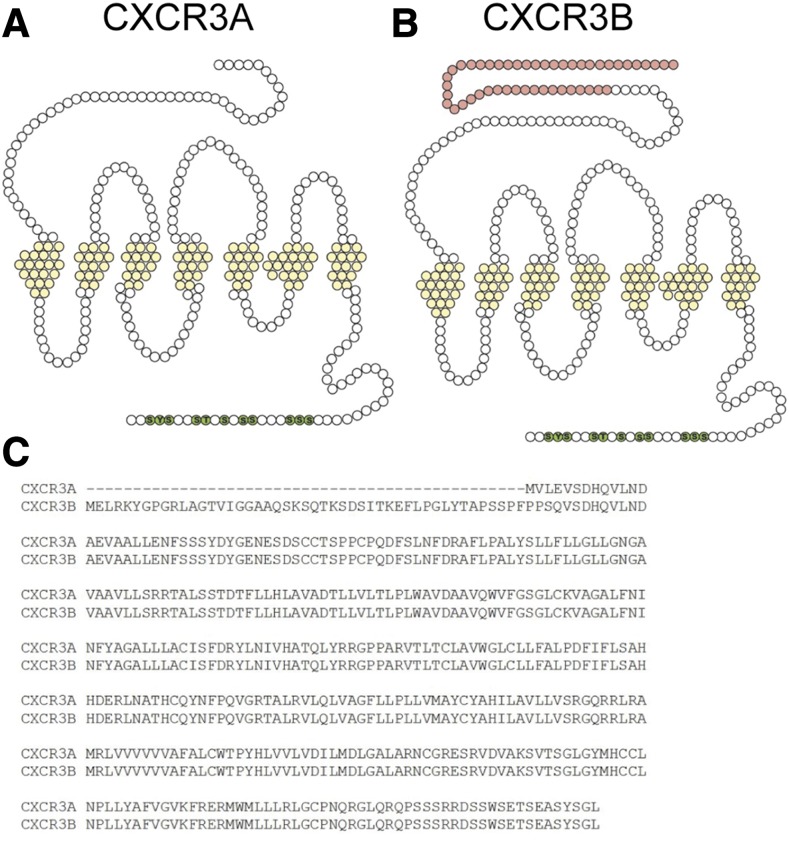
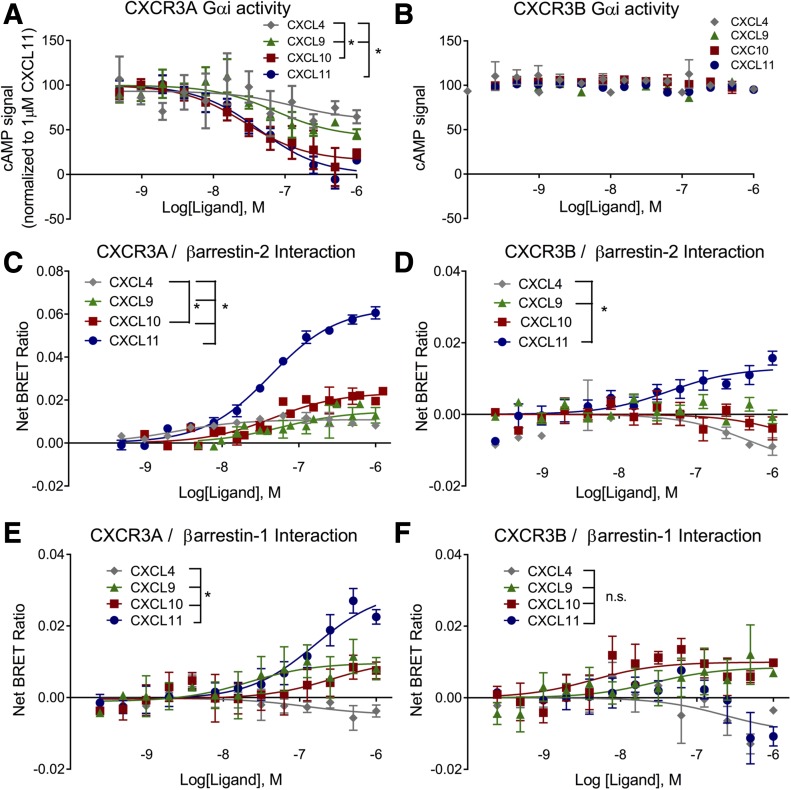
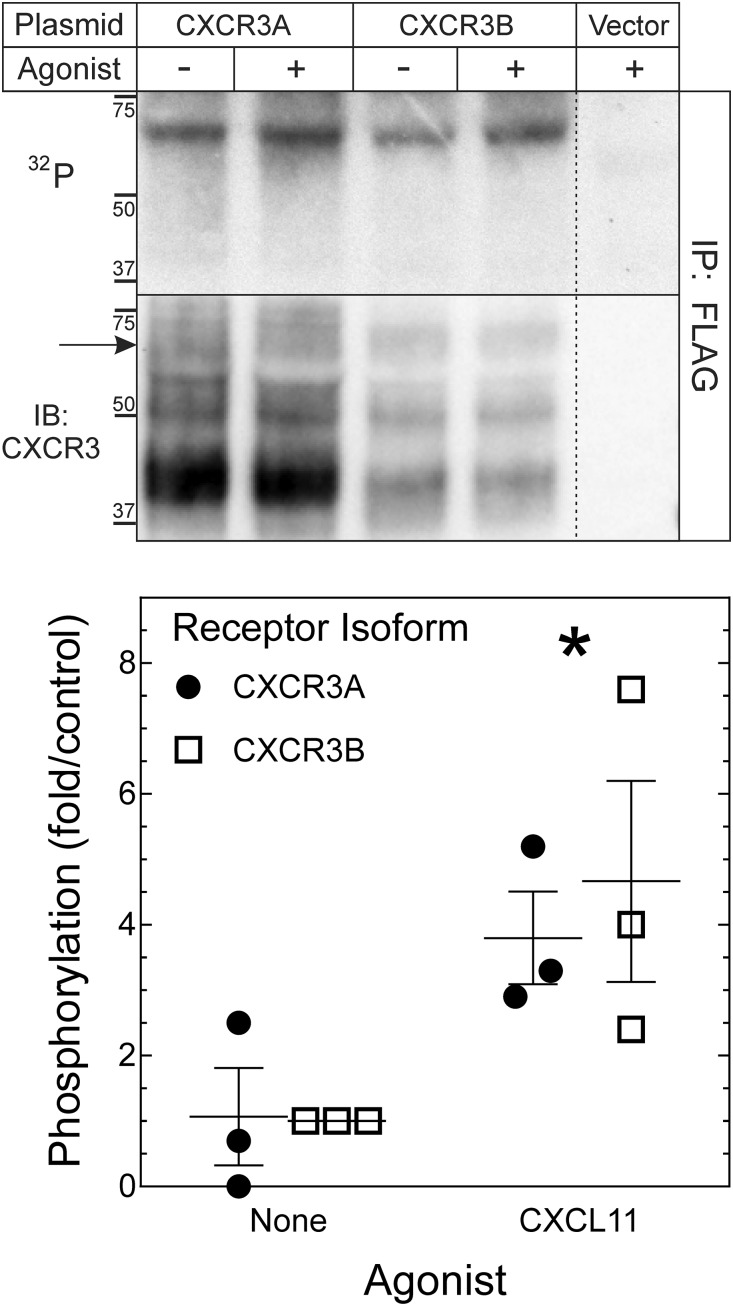
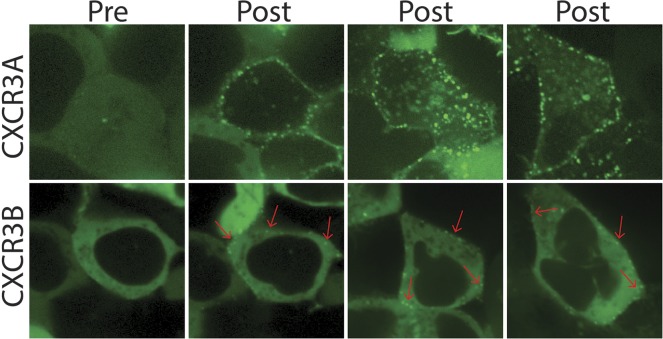
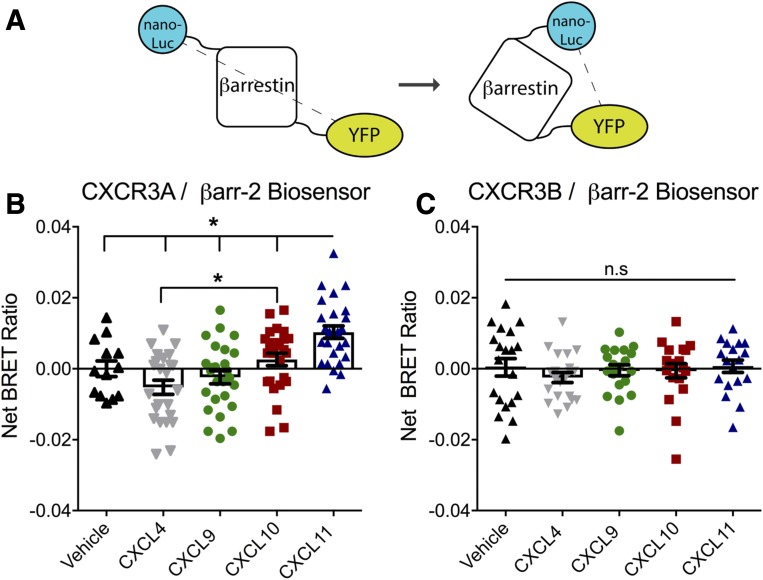
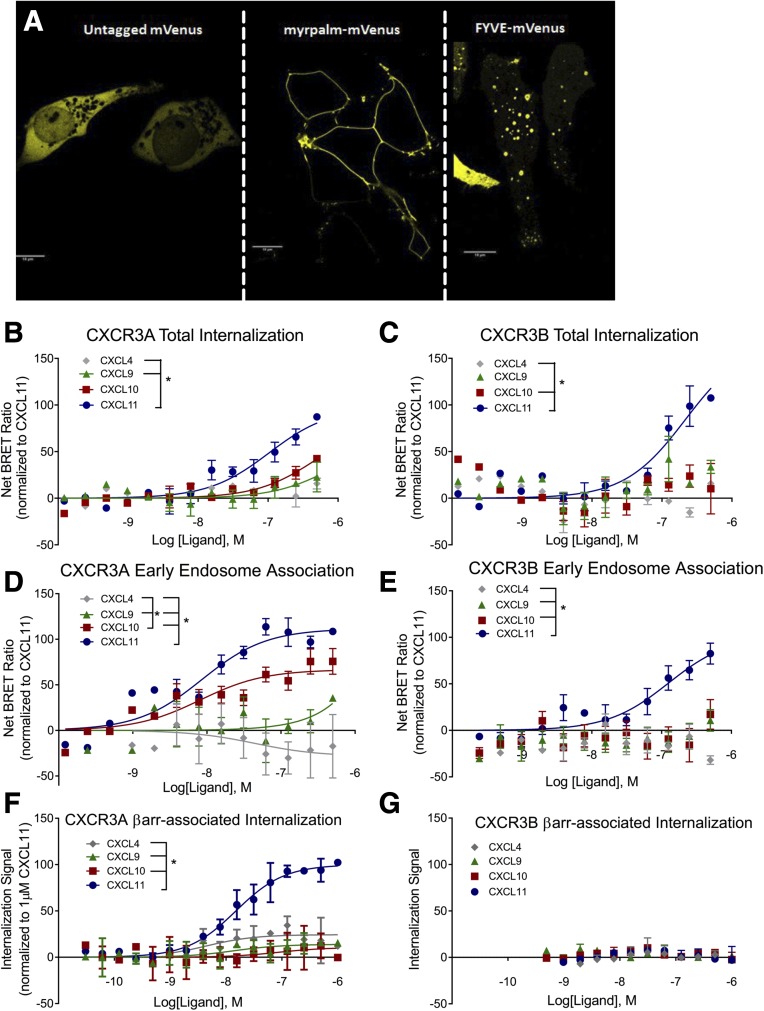
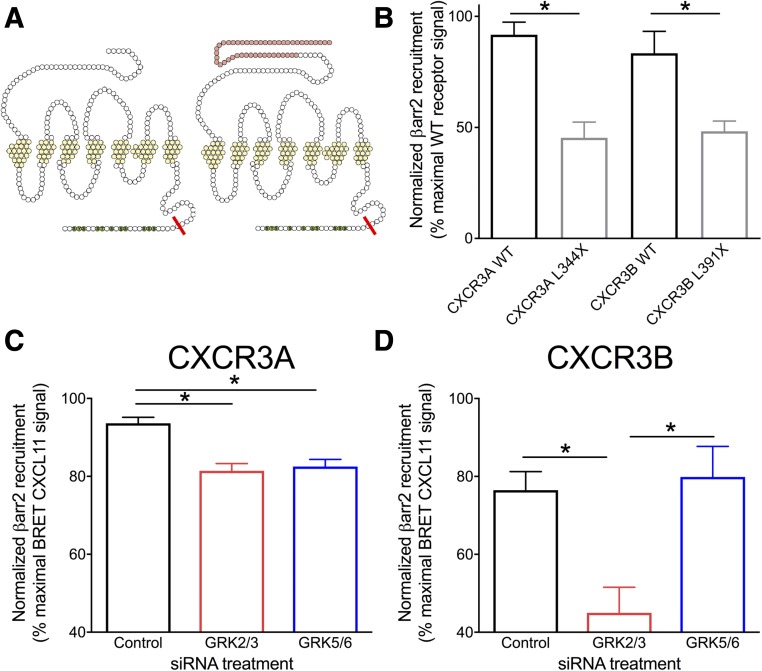
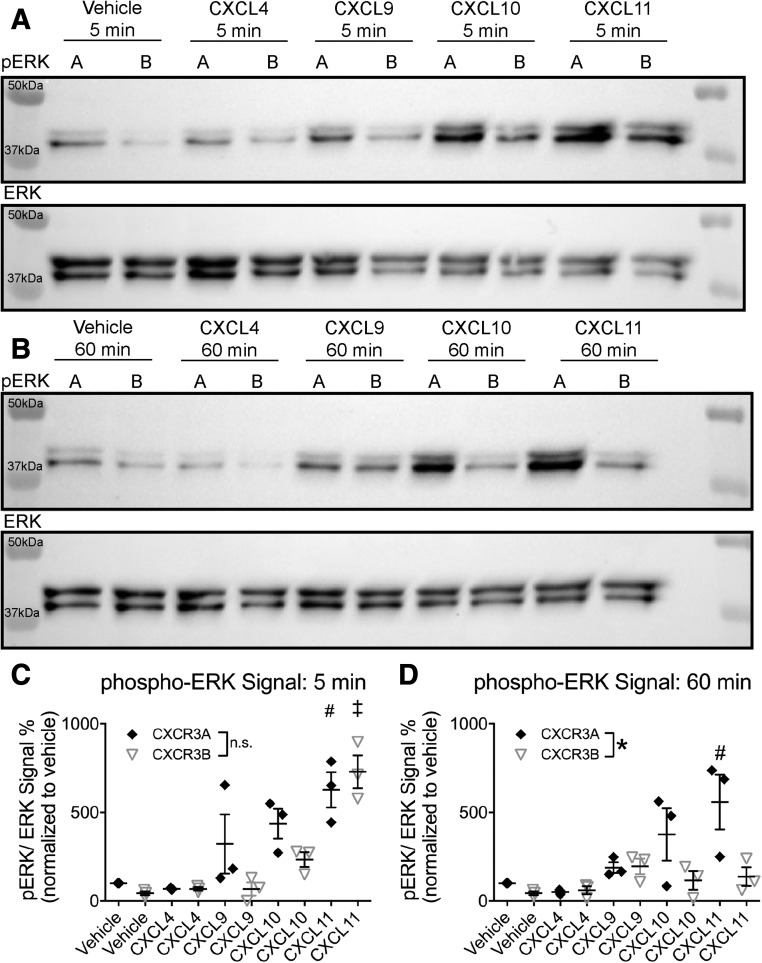
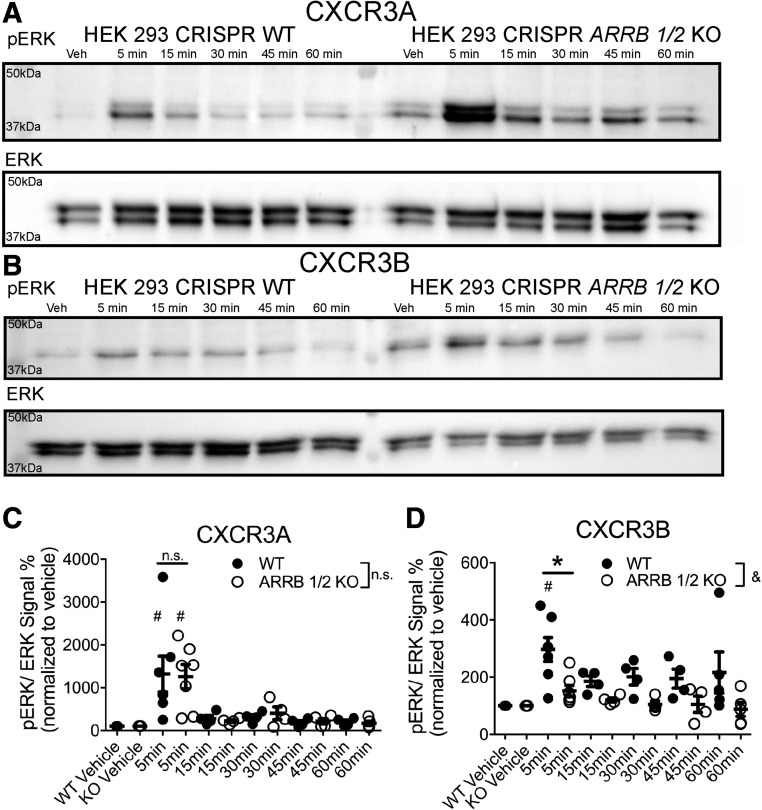
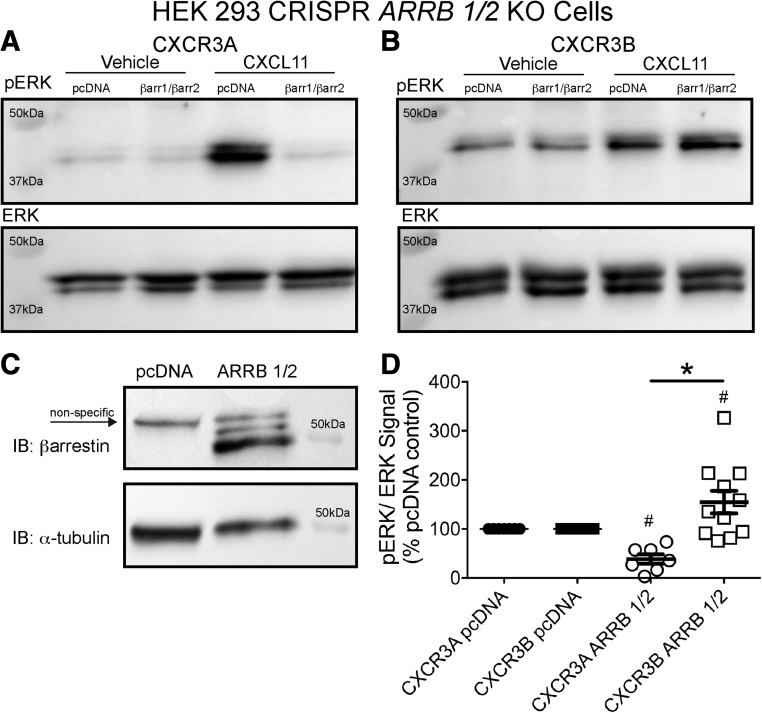
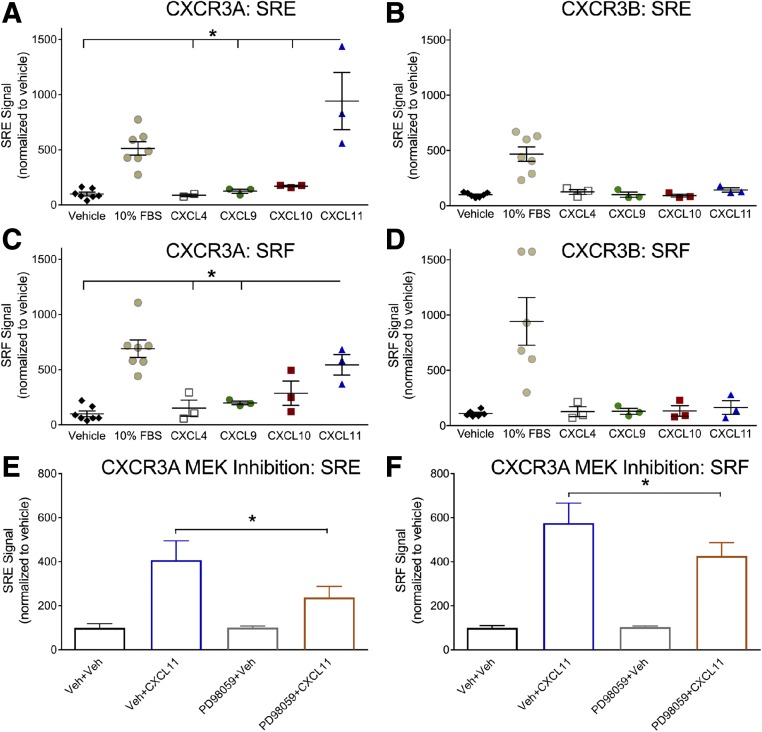
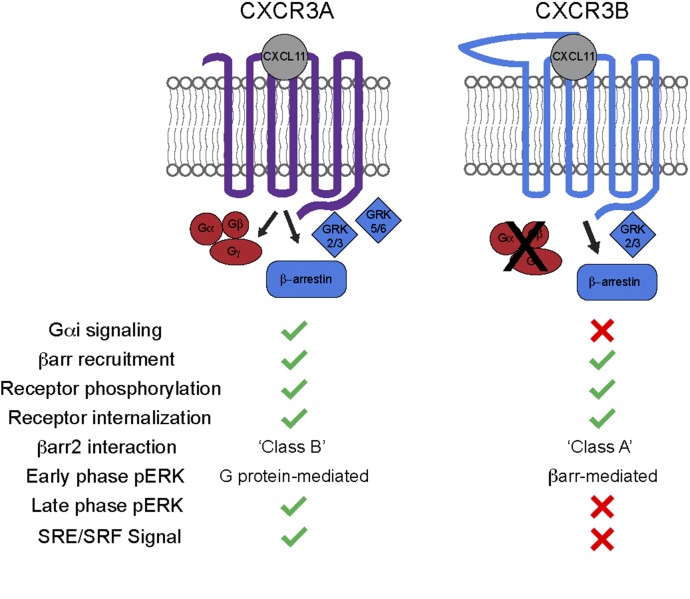
Biased agonism, the ability of different ligands for the same receptor to selectively activate some signaling pathways while blocking others, is now an established paradigm for G protein-coupled receptor signaling. One group of receptors in which endogenous bias is critical is the chemokine system, consisting of over 50 ligands and 20 receptors that bind one another with significant promiscuity. We have previously demonstrated that ligands for the same receptor can cause biased signaling responses. The goal of this study was to identify mechanisms that could underlie biased signaling between different receptor splice variants. The C-X-C motif chemokine receptor 3 (CXCR3) has two splice variants, CXCR3A and CXCR3B, which differ by 51 amino acids at its N-terminus. Consistent with an earlier study, we found that C-X-C motif chemokine ligands 4, 9, 10, and 11 all activated G αi at CXCR3A, while at CXCR3B these ligands demonstrated no measurable G αi or G αs activity. β-arrestin (βarr) was recruited at a reduced level to CXCR3B relative to CXCR3A, which was also associated with differences in βarr2 conformation. βarr2 recruitment to CXCR3A was attenuated by both G protein receptor kinase (GRK) 2/3 and GRK5/6 knockdown, while only GRK2/3 knockdown blunted recruitment to CXCR3B. Extracellular regulated kinase 1/2 phosphorylation downstream from CXCR3A and CXCR3B was increased and decreased, respectively, by βarr1/2 knockout. The splice variants also differentially activated transcriptional reporters. These findings demonstrate that differential splicing of CXCR3 results in biased responses associated with distinct patterns of βarr conformation and recruitment. Differential splicing may serve as a common mechanism for generating biased signaling and provides insights into how chemokine receptor signaling can be modulated post-transcriptionally.
Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
References
-
- Ahn S, Kim J, Lucaveche CL, Reedy MC, Luttrell LM, Lefkowitz RJ, Daaka Y. (2002) Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J Biol Chem 277:26642–26651. - PubMed
-
- Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. (1992) β-Arrestin2, a novel member of the arrestin/β-arrestin gene family. J Biol Chem 267:17882–17890. - PubMed
-
- Berchiche YA, Sakmar TP. (2016) CXC chemokine receptor 3 alternative splice variants selectively activate different signaling pathways. Mol Pharmacol 90:483–495. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
