

IL-17 induced NOTCH1 activation in oligodendrocyte progenitor cells enhances proliferation and inflammatory gene expression
- PMID: 28561022
- PMCID: PMC5460031
- DOI: 10.1038/ncomms15508
IL-17 induced NOTCH1 activation in oligodendrocyte progenitor cells enhances proliferation and inflammatory gene expression
Abstract
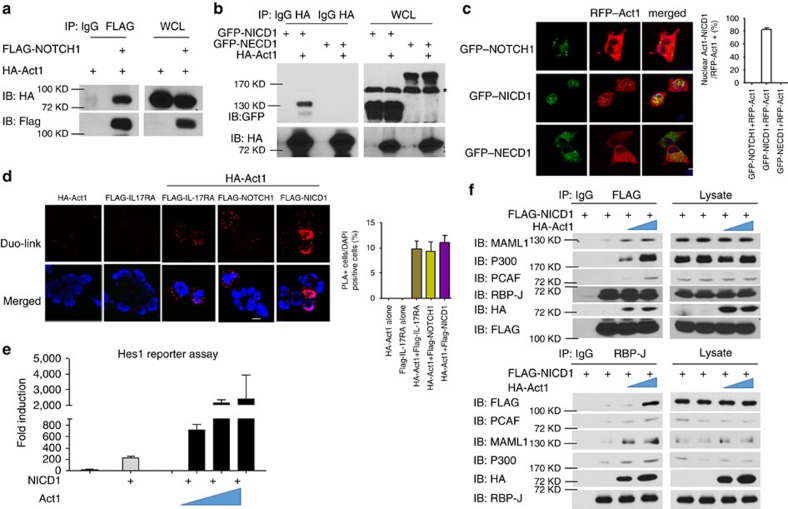
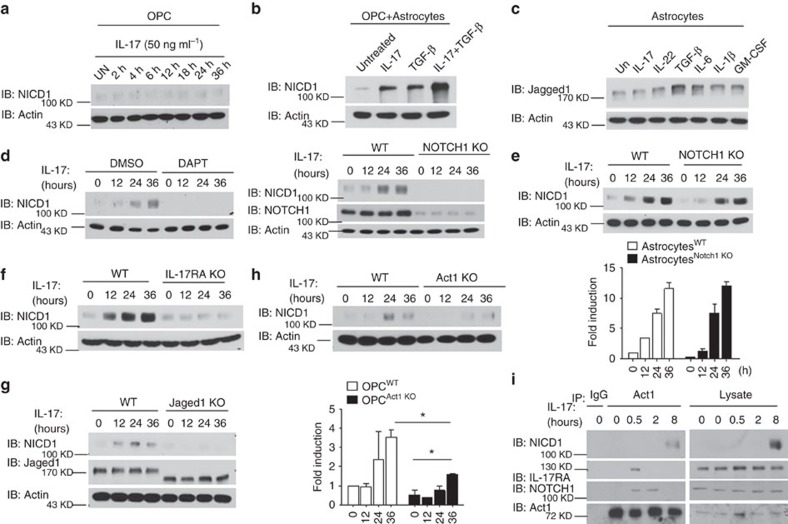
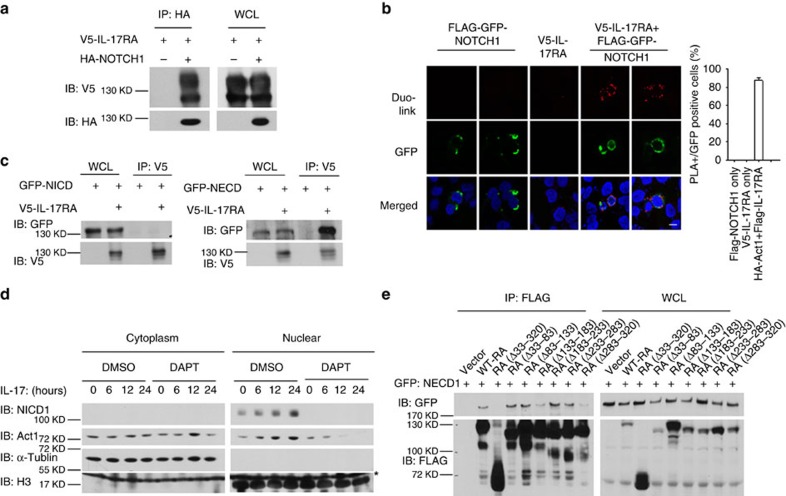
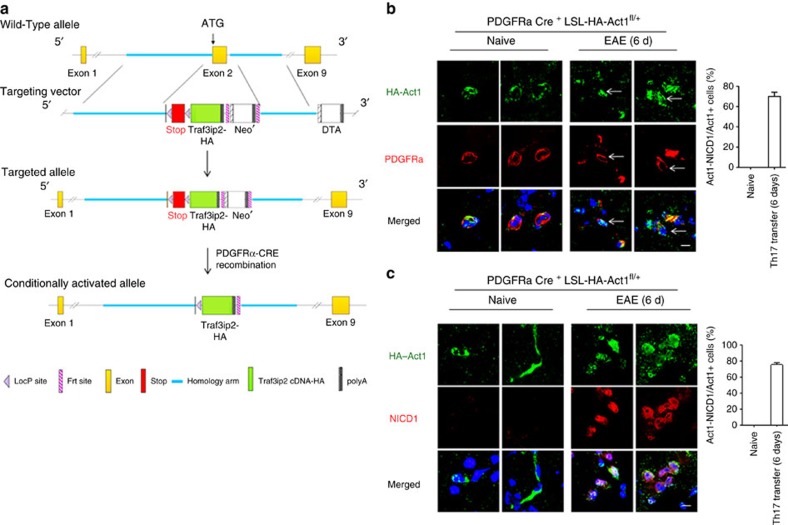
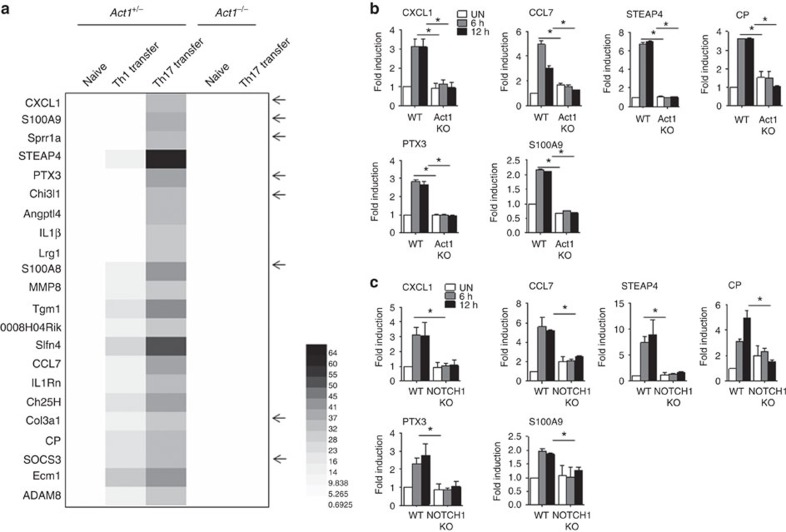
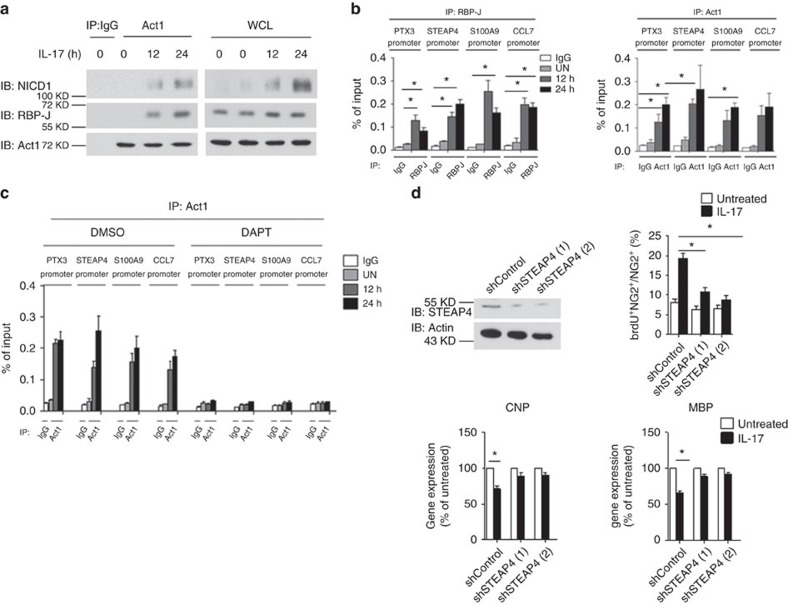
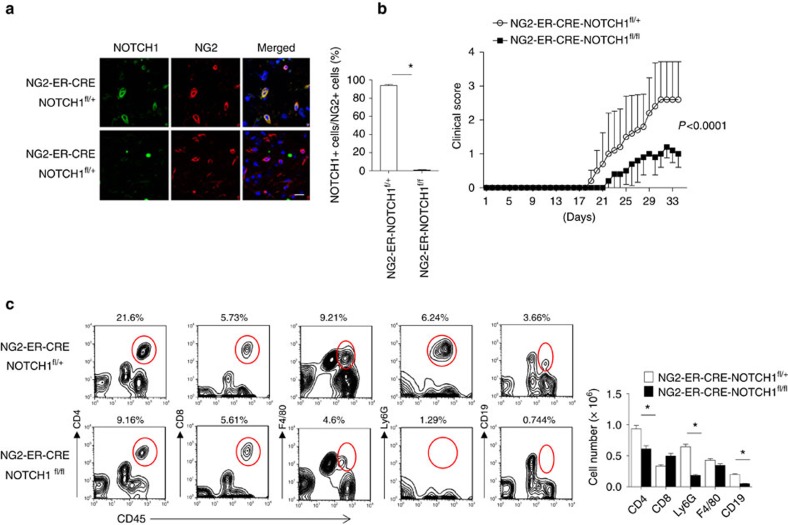
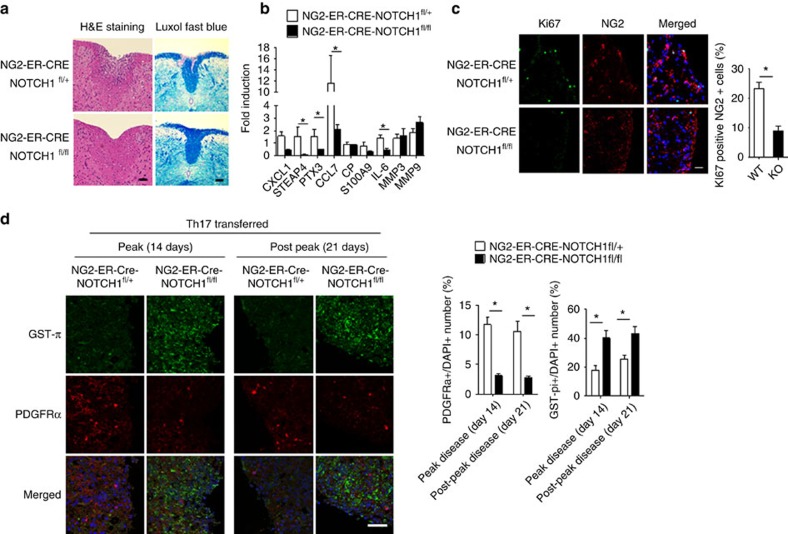
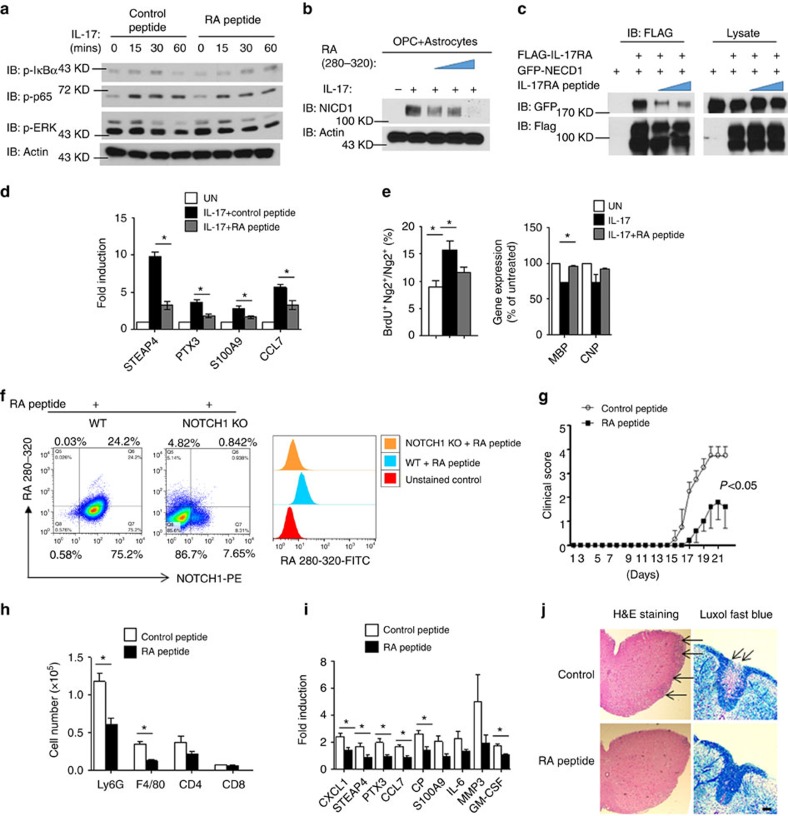
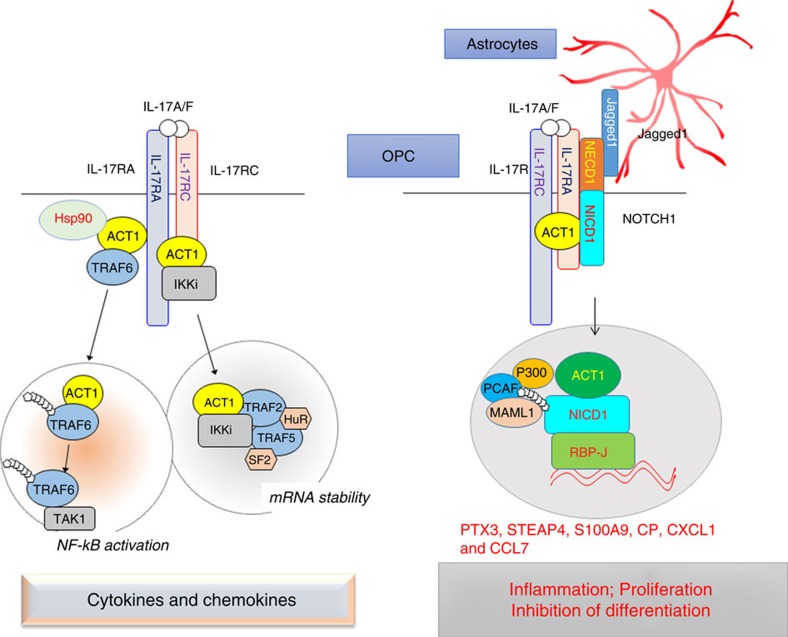
NOTCH1 signalling contributes to defective remyelination by impairing differentiation of oligodendrocyte progenitor cells (OPCs). Here we report that IL-17 stimulation induces NOTCH1 activation in OPCs, contributing to Th17-mediated demyelinating disease. Mechanistically, IL-17R interacts with NOTCH1 via the extracellular domain, which facilitates the cleavage of NOTHC1 intracellular domain (NICD1). IL-17-induced NOTCH1 activation results in the interaction of IL-17R adaptor Act1 with NICD1, followed by the translocation of the Act1-NICD1 complex into the nucleus. Act1-NICD1 are recruited to the promoters of several NOTCH1 target genes (including STEAP4, a metalloreductase important for inflammation and cell proliferation) that are specifically induced in the spinal cord by Th17 cells. A decoy peptide disrupting the IL-17RA-NOTCH1 interaction inhibits IL-17-induced NOTCH1 activation and attenuates Th17-mediated experimental autoimmune encephalitis (EAE). Taken together, these findings demonstrate critical crosstalk between the IL-17 and NOTCH1 pathway, regulating Th17-induced inflammatory and proliferative genes to promote demyelinating disease.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Becher B., Bechmann I. & Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J. Mol. Med. 84, 532–543 (2006). - PubMed
-
- Bar-Or A. Immunology of multiple sclerosis. Neurol. Clin. 23, 149–175 (2005). - PubMed
-
- Steinman L. Multiple sclerosis: a two-stage disease. Nat. Immunol. 2, 762–764 (2001). - PubMed
-
- Stromnes I. M. & Goverman J. M. Active induction of experimental allergic encephalomyelitis. Nat. Protoc. 1, 1810–1819 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
