

Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory
- PMID: 28562591
- PMCID: PMC5505514
- DOI: 10.1038/nature22405
Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory
Abstract
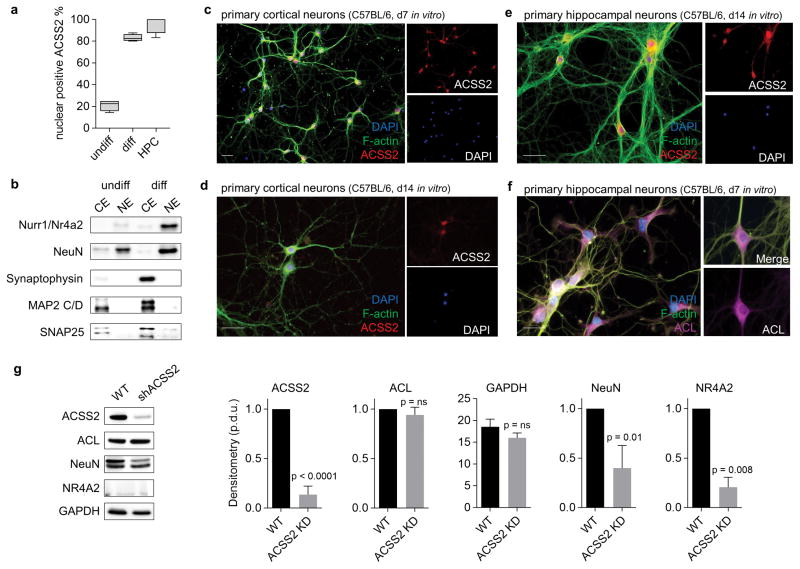
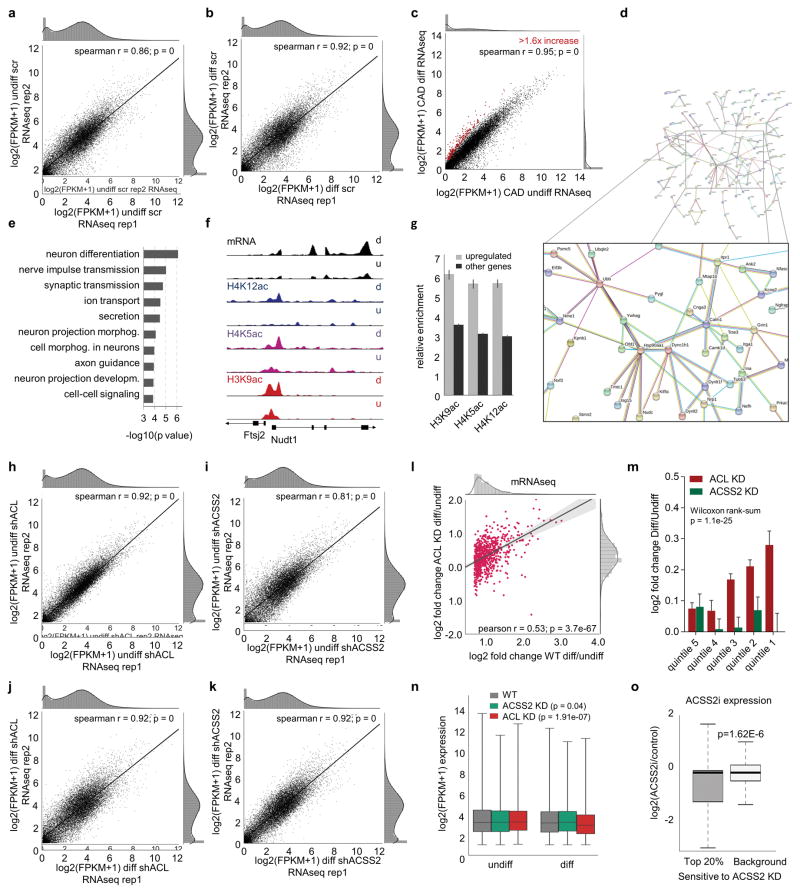
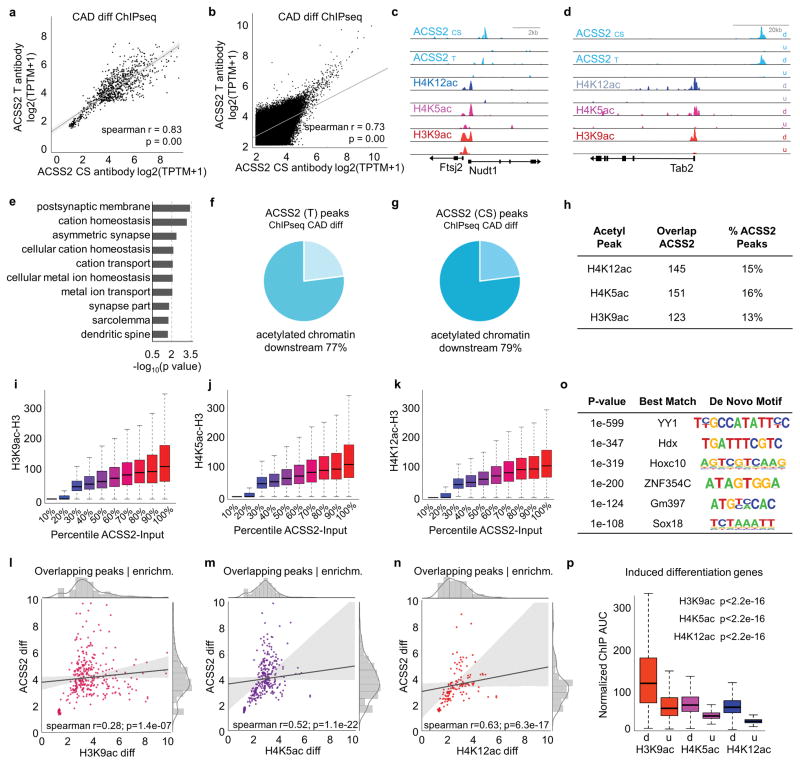
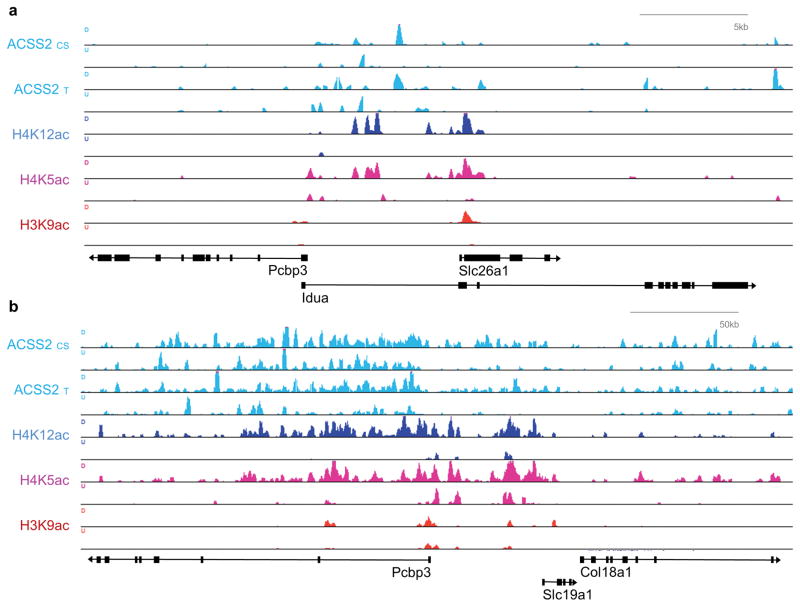
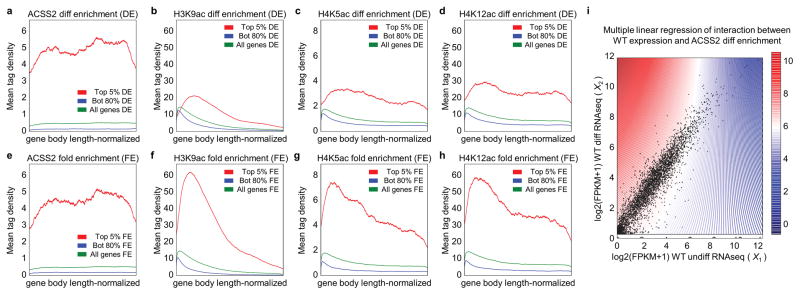
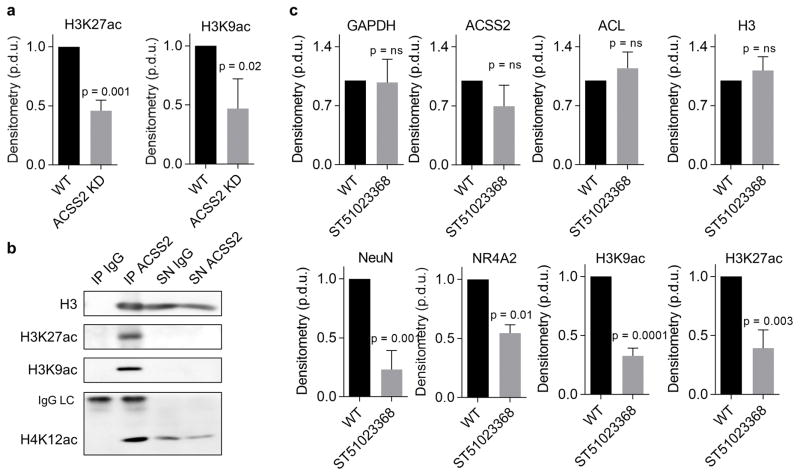
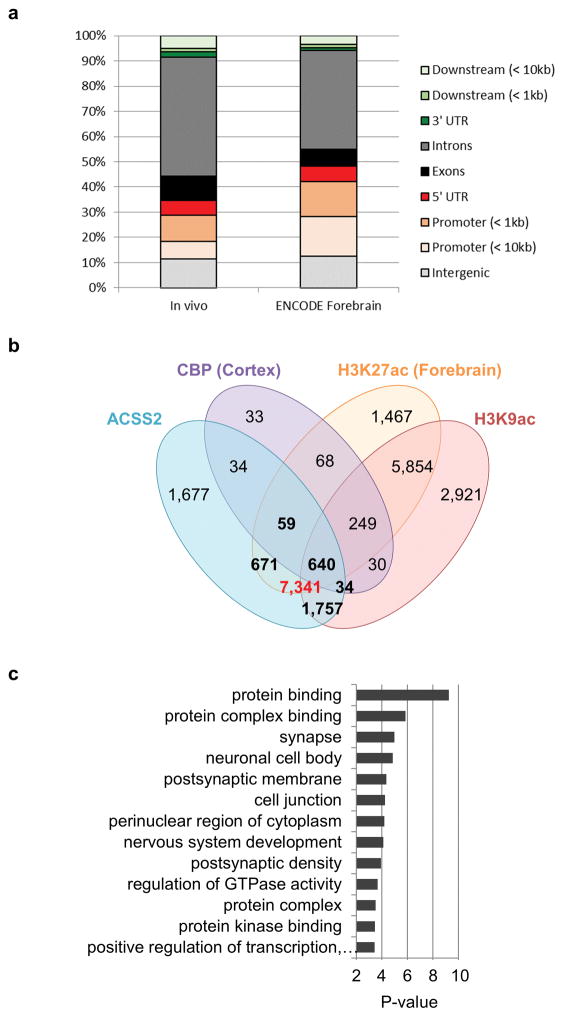
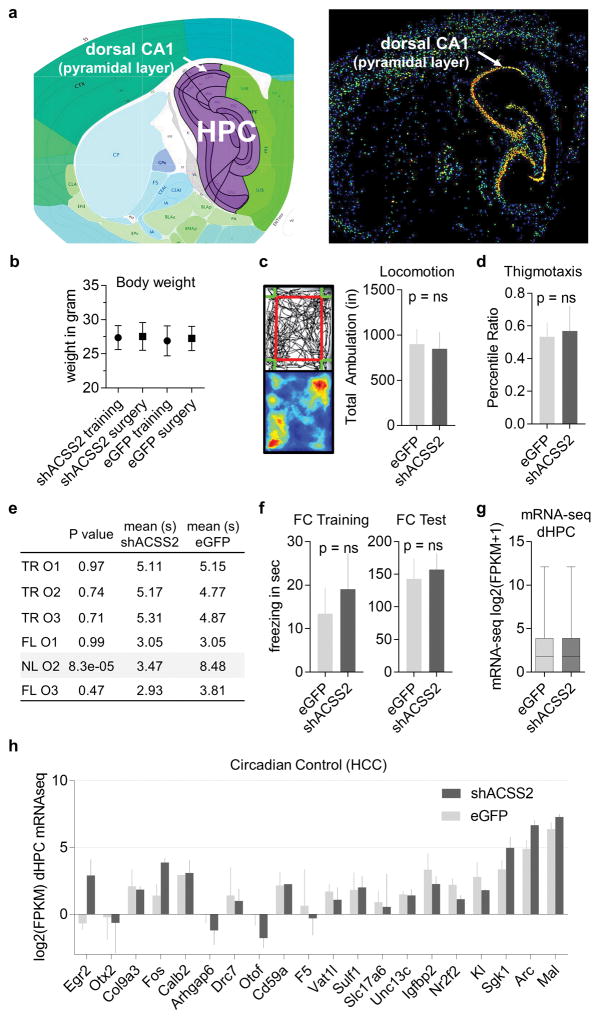
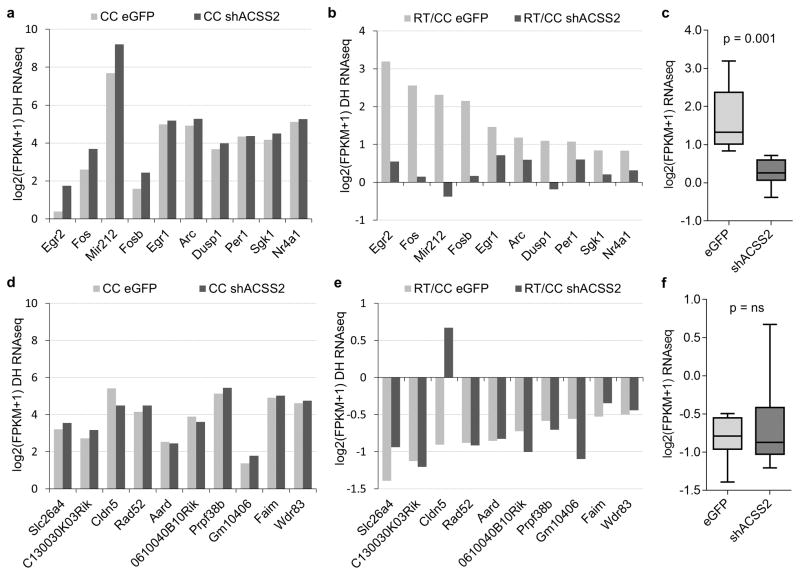
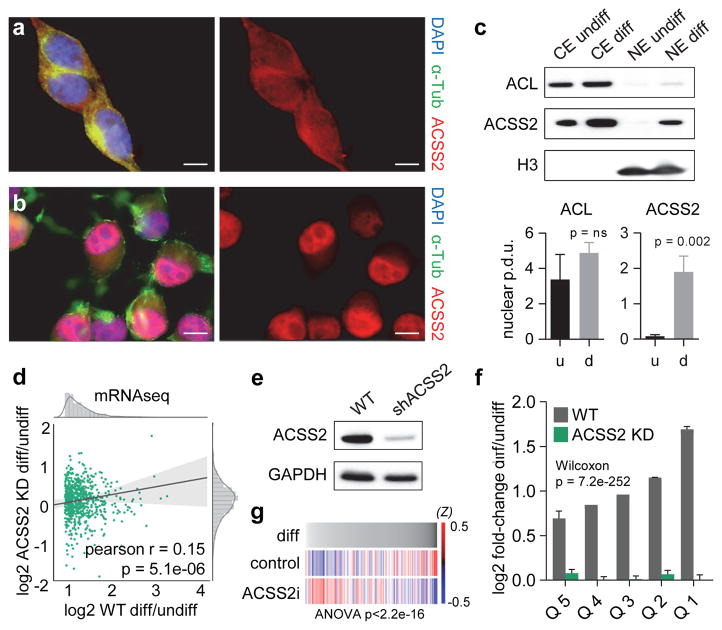
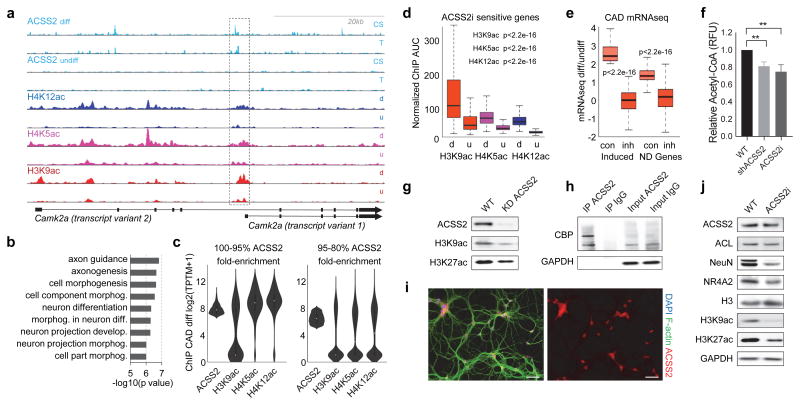
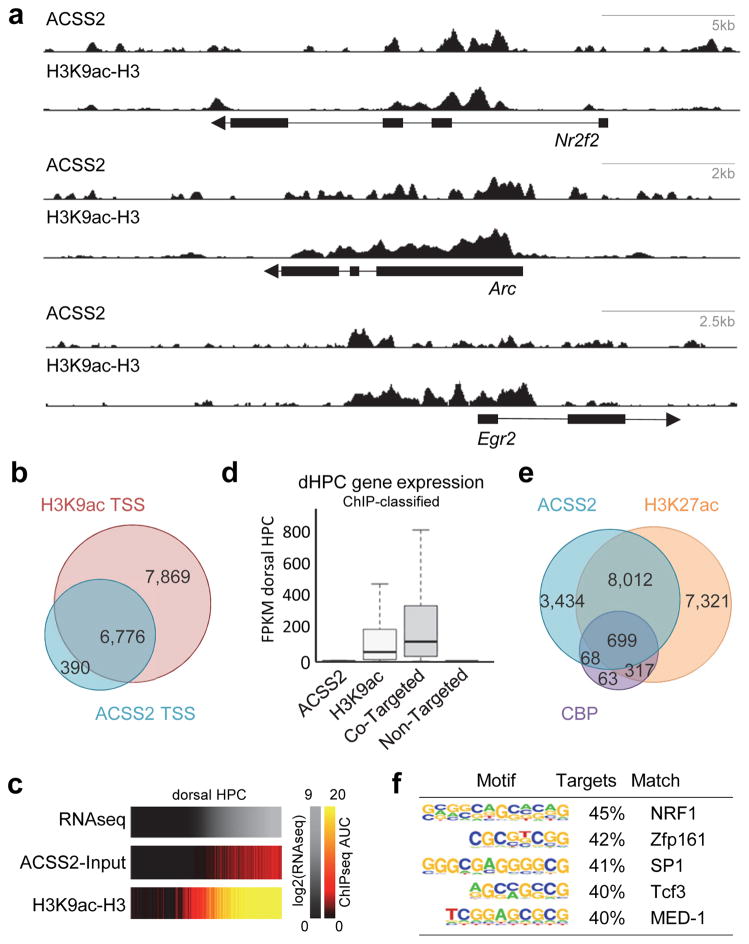
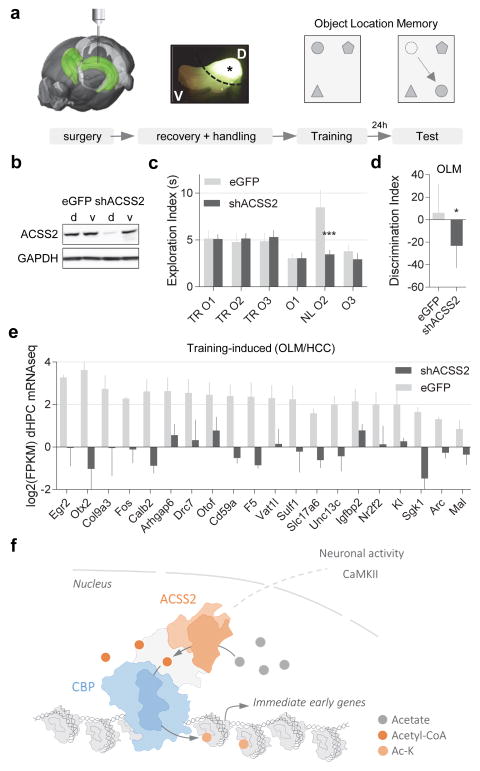
Metabolic production of acetyl coenzyme A (acetyl-CoA) is linked to histone acetylation and gene regulation, but the precise mechanisms of this process are largely unknown. Here we show that the metabolic enzyme acetyl-CoA synthetase 2 (ACSS2) directly regulates histone acetylation in neurons and spatial memory in mammals. In a neuronal cell culture model, ACSS2 increases in the nuclei of differentiating neurons and localizes to upregulated neuronal genes near sites of elevated histone acetylation. A decrease in ACSS2 lowers nuclear acetyl-CoA levels, histone acetylation, and responsive expression of the cohort of neuronal genes. In adult mice, attenuation of hippocampal ACSS2 expression impairs long-term spatial memory, a cognitive process that relies on histone acetylation. A decrease in ACSS2 in the hippocampus also leads to defective upregulation of memory-related neuronal genes that are pre-bound by ACSS2. These results reveal a connection between cellular metabolism, gene regulation, and neural plasticity and establish a link between acetyl-CoA generation 'on-site' at chromatin for histone acetylation and the transcription of key neuronal genes.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Molecular biology: Local metabolites linked to memory.Nature. 2017 Jun 15;546(7658):361-362. doi: 10.1038/nature22498. Epub 2017 May 31. Nature. 2017. PMID: 28562586 No abstract available.
-
Gene expression: ACSS2 boosts local histone acetylation.Nat Rev Mol Cell Biol. 2017 Jul;18(7):405. doi: 10.1038/nrm.2017.61. Epub 2017 Jun 7. Nat Rev Mol Cell Biol. 2017. PMID: 28588257 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
