

A Continuum Poisson-Boltzmann Model for Membrane Channel Proteins
- PMID: 28564540
- PMCID: PMC5728381
- DOI: 10.1021/acs.jctc.7b00382
A Continuum Poisson-Boltzmann Model for Membrane Channel Proteins
Abstract
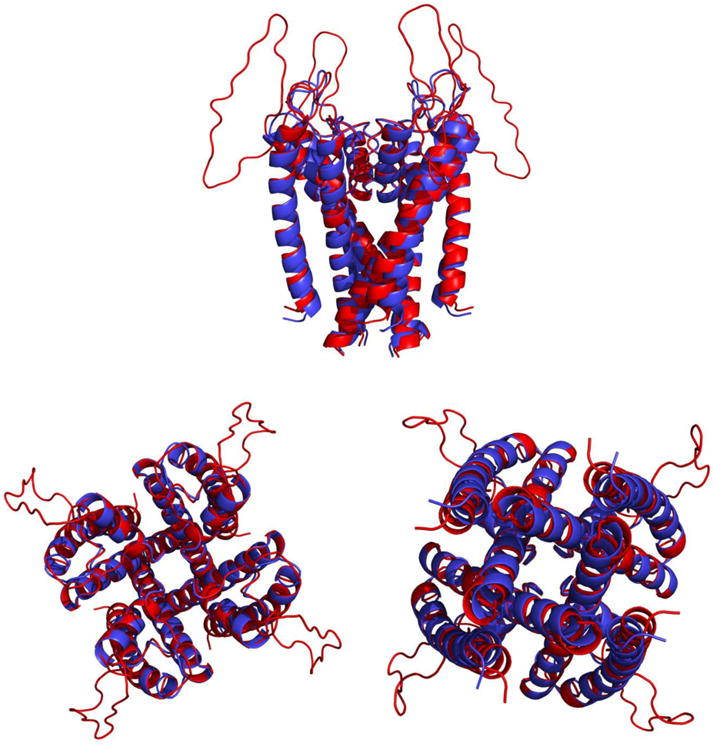
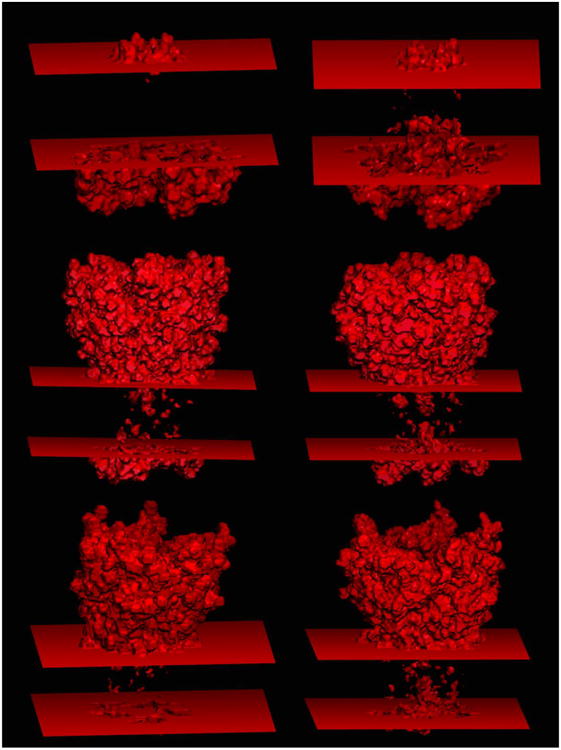
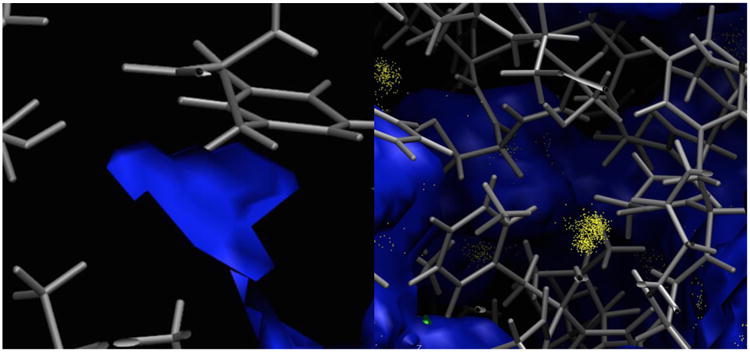
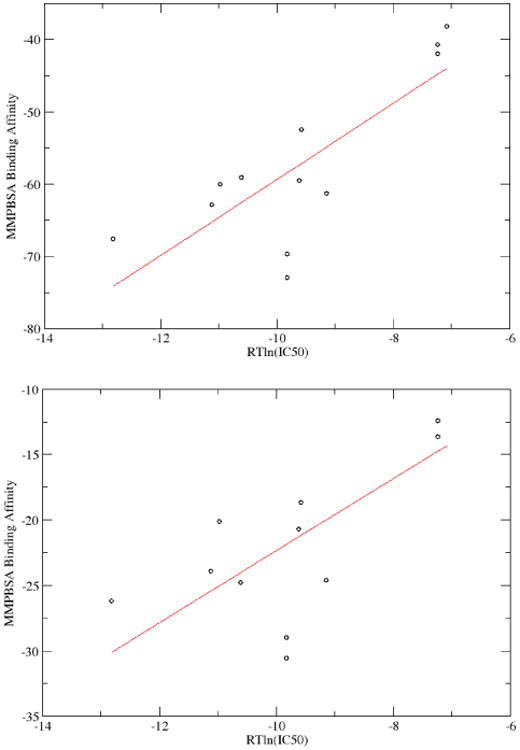
Membrane proteins constitute a large portion of the human proteome and perform a variety of important functions as membrane receptors, transport proteins, enzymes, signaling proteins, and more. Computational studies of membrane proteins are usually much more complicated than those of globular proteins. Here, we propose a new continuum model for Poisson-Boltzmann calculations of membrane channel proteins. Major improvements over the existing continuum slab model are as follows: (1) The location and thickness of the slab model are fine-tuned based on explicit-solvent MD simulations. (2) The highly different accessibilities in the membrane and water regions are addressed with a two-step, two-probe grid-labeling procedure. (3) The water pores/channels are automatically identified. The new continuum membrane model is optimized (by adjusting the membrane probe, as well as the slab thickness and center) to best reproduce the distributions of buried water molecules in the membrane region as sampled in explicit water simulations. Our optimization also shows that the widely adopted water probe of 1.4 Å for globular proteins is a very reasonable default value for membrane protein simulations. It gives the best compromise in reproducing the explicit water distributions in membrane channel proteins, at least in the water accessible pore/channel regions. Finally, we validate the new membrane model by carrying out binding affinity calculations for a potassium channel, and we observe good agreement with the experimental results.
Figures


Similar articles
-
A New Poisson-Nernst-Planck Model with Ion-Water Interactions for Charge Transport in Ion Channels.Bull Math Biol. 2016 Aug;78(8):1703-26. doi: 10.1007/s11538-016-0196-7. Epub 2016 Aug 1. Bull Math Biol. 2016. PMID: 27480225
-
Quantum dynamics in continuum for proton transport II: Variational solvent-solute interface.Int J Numer Method Biomed Eng. 2012 Jan;28(1):25-51. doi: 10.1002/cnm.1458. Epub 2011 Aug 9. Int J Numer Method Biomed Eng. 2012. PMID: 22328970 Free PMC article.
-
A Finite Element Solution of Lateral Periodic Poisson-Boltzmann Model for Membrane Channel Proteins.Int J Mol Sci. 2018 Feb 28;19(3):695. doi: 10.3390/ijms19030695. Int J Mol Sci. 2018. PMID: 29495644 Free PMC article.
-
Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations.Biochim Biophys Acta Biomembr. 2018 Apr;1860(4):909-926. doi: 10.1016/j.bbamem.2017.10.033. Epub 2017 Nov 4. Biochim Biophys Acta Biomembr. 2018. PMID: 29113819 Review.
-
Models and simulations of ion channels and related membrane proteins.Curr Opin Struct Biol. 1998 Apr;8(2):237-44. doi: 10.1016/s0959-440x(98)80045-6. Curr Opin Struct Biol. 1998. PMID: 9631299 Review.
Cited by
-
Estimating the Roles of Protonation and Electronic Polarization in Absolute Binding Affinity Simulations.J Chem Theory Comput. 2021 Apr 13;17(4):2541-2555. doi: 10.1021/acs.jctc.0c01305. Epub 2021 Mar 25. J Chem Theory Comput. 2021. PMID: 33764050 Free PMC article.
-
Recent Developments and Applications of the MMPBSA Method.Front Mol Biosci. 2018 Jan 10;4:87. doi: 10.3389/fmolb.2017.00087. eCollection 2017. Front Mol Biosci. 2018. PMID: 29367919 Free PMC article. Review.
-
Novel compounds that specifically bind and modulate MscL: insights into channel gating mechanisms.FASEB J. 2019 Mar;33(3):3180-3189. doi: 10.1096/fj.201801628R. Epub 2018 Oct 25. FASEB J. 2019. PMID: 30359098 Free PMC article.
-
DelPhi Suite: New Developments and Review of Functionalities.J Comput Chem. 2019 Oct 30;40(28):2502-2508. doi: 10.1002/jcc.26006. Epub 2019 Jun 25. J Comput Chem. 2019. PMID: 31237360 Free PMC article.
-
GraphDeep-hERG: Graph Neural Network PharmacoAnalytics for Assessing hERG-Related Cardiotoxicity.Pharm Res. 2025 Apr;42(4):579-591. doi: 10.1007/s11095-025-03848-w. Epub 2025 Mar 26. Pharm Res. 2025. PMID: 40140128
References
-
- Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–1126. - PubMed
-
- Overington JP, Al-Lazikani B, Hopkins AL. Opinion - How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. - PubMed
-
- Davis ME, Mccammon JA. Electrostatics in Biomolecular Structure and Dynamics. Chem Rev. 1990;90:509–521.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources