

Effect of Human Genetic Variability on Gene Expression in Dorsal Root Ganglia and Association with Pain Phenotypes
- PMID: 28564610
- PMCID: PMC5524461
- DOI: 10.1016/j.celrep.2017.05.018
Effect of Human Genetic Variability on Gene Expression in Dorsal Root Ganglia and Association with Pain Phenotypes
Abstract
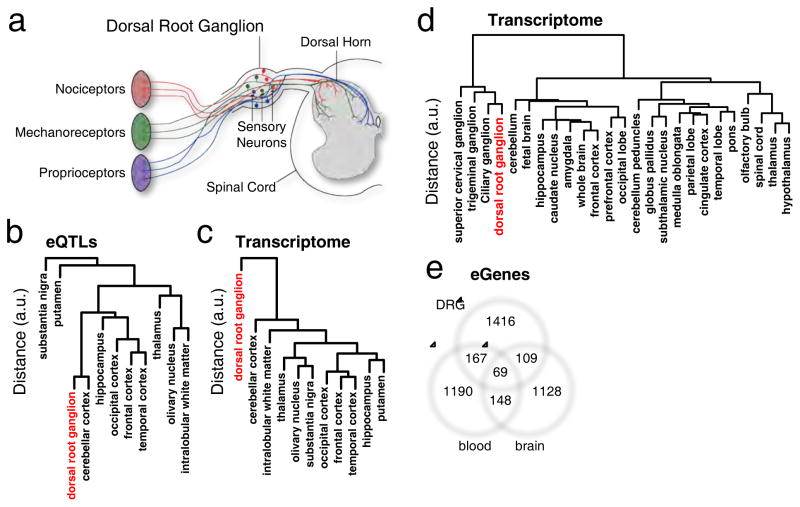
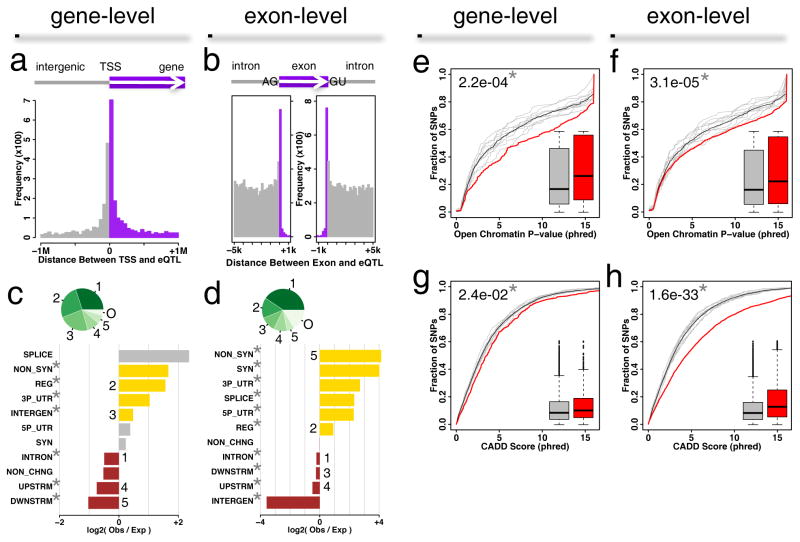
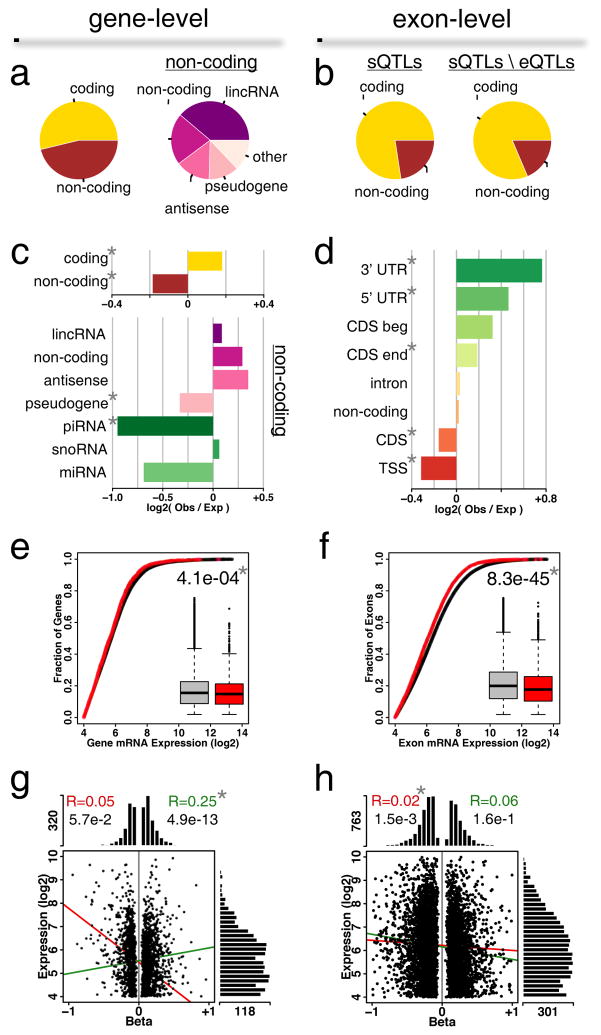
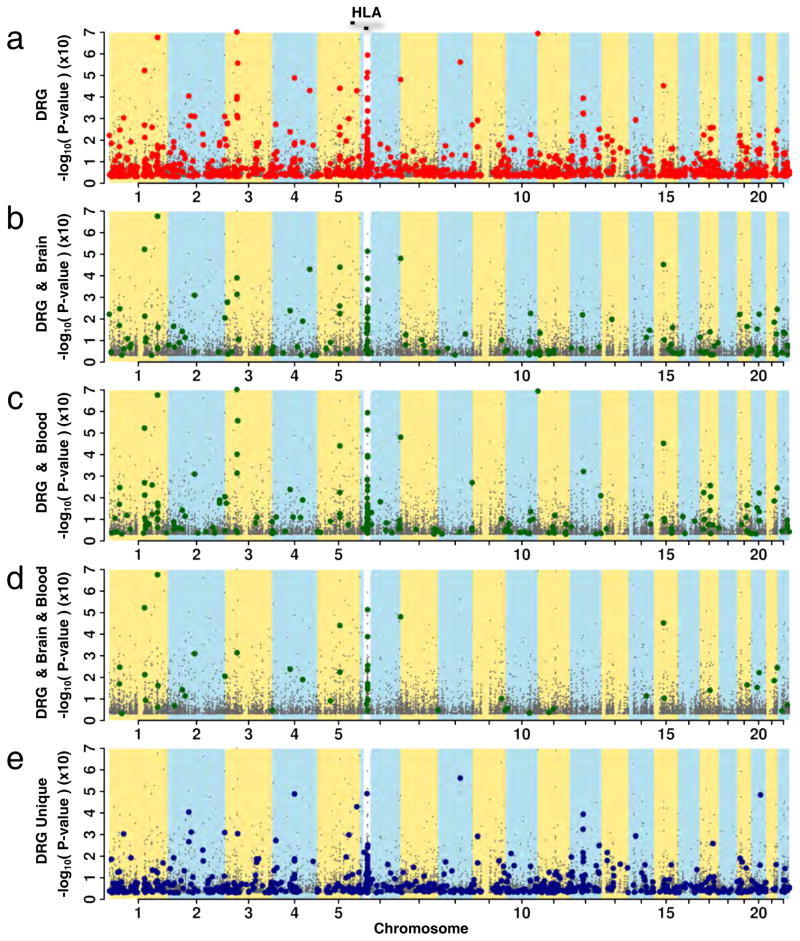
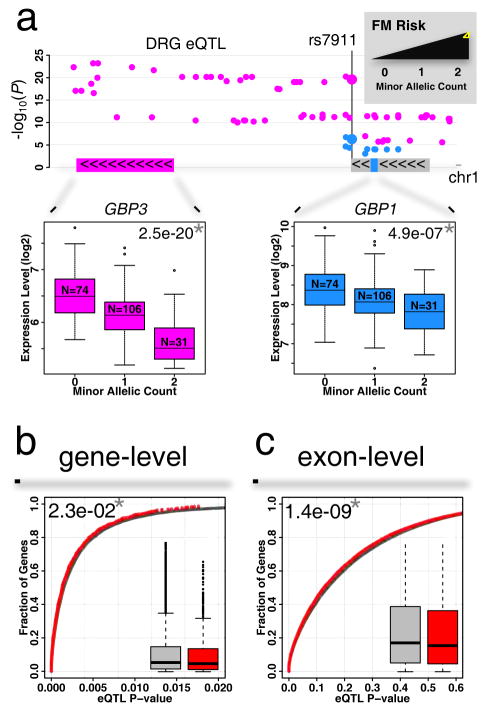
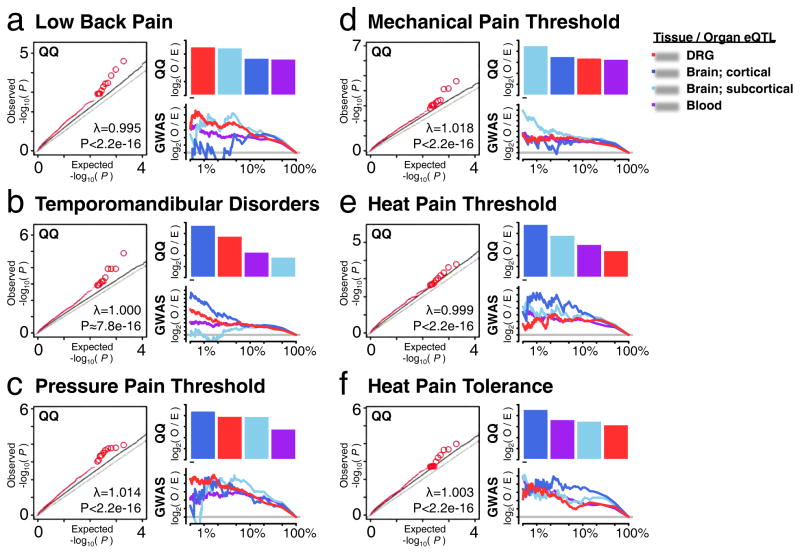
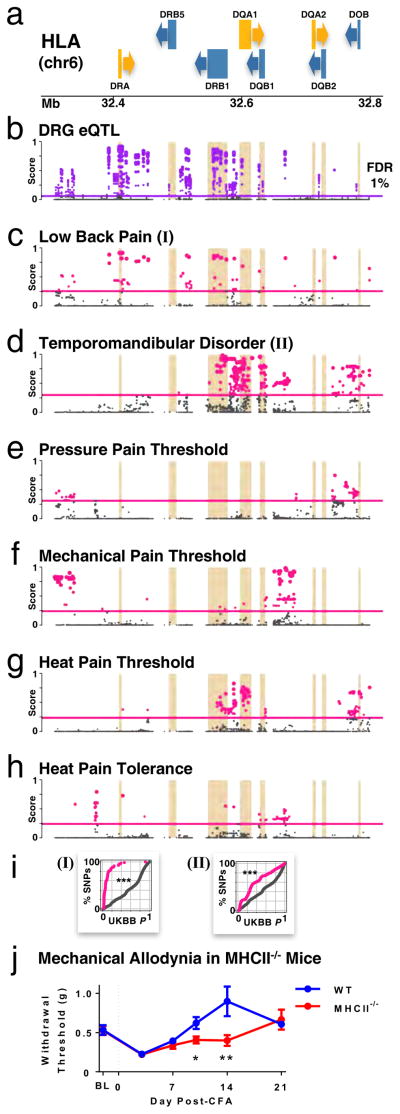
Dorsal root ganglia (DRG) relay sensory information to the brain, giving rise to the perception of pain, disorders of which are prevalent and burdensome. Here, we mapped expression quantitative trait loci (eQTLs) in a collection of human DRGs. DRG eQTLs were enriched within untranslated regions of coding genes of low abundance, with some overlapping with other brain regions and blood cell cis-eQTLs. We confirm functionality of identified eQTLs through their significant enrichment within open chromatin and highly deleterious SNPs, particularly at the exon level, suggesting substantial contribution of eQTLs to alternative splicing regulation. We illustrate pain-related genetic association results explained by DRG eQTLs, with the strongest evidence for contribution of the human leukocyte antigen (HLA) locus, confirmed using a mouse inflammatory pain model. Finally, we show that DRG eQTLs are found among hits in numerous genome-wide association studies, suggesting that this dataset will help address pain components of non-pain disorders.
Keywords: DRG; GWAS; QST; SNPs; dorsal root ganglion; eQTLs; expression quantitative trait loci; genome-wide association study; pain; quantitative sensory testing; single nucleotide polymorphism.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
