

Discovery and Mechanistic Study of Benzamide Derivatives That Modulate Hepatitis B Virus Capsid Assembly
- PMID: 28566379
- PMCID: PMC5533917
- DOI: 10.1128/JVI.00519-17
Discovery and Mechanistic Study of Benzamide Derivatives That Modulate Hepatitis B Virus Capsid Assembly
Abstract
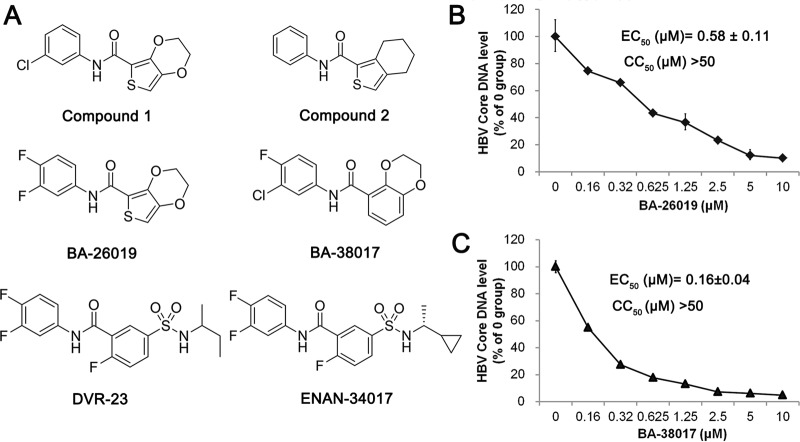
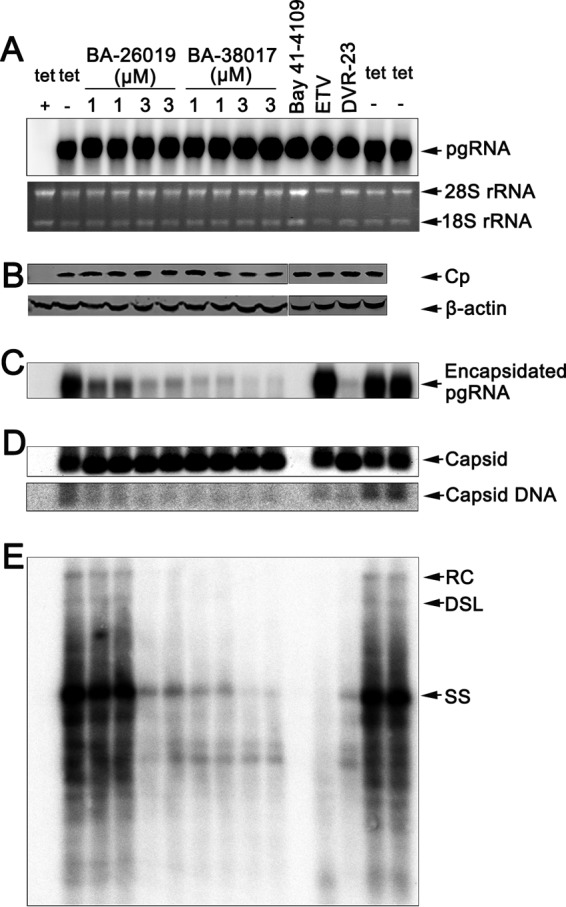
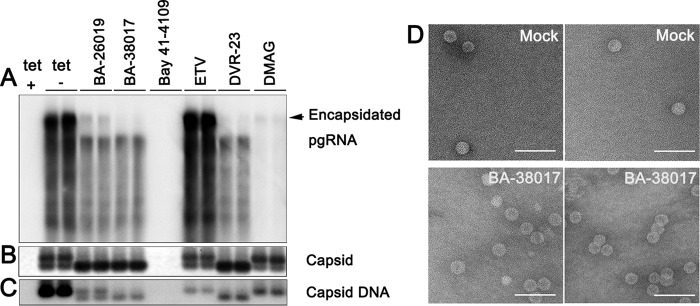
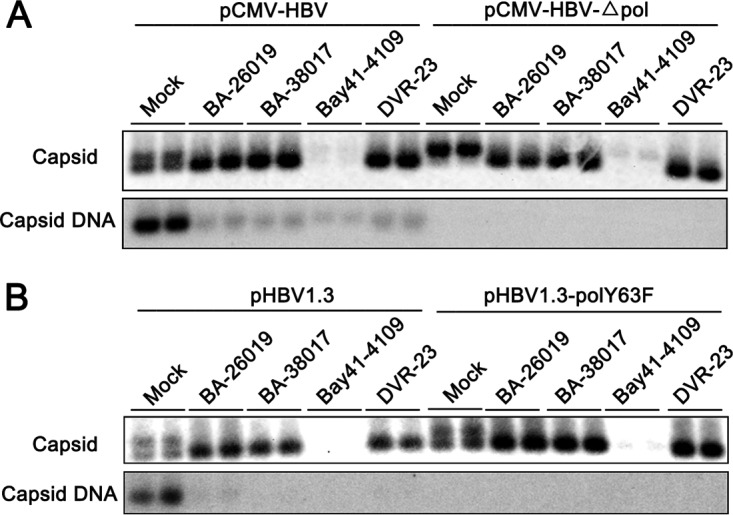
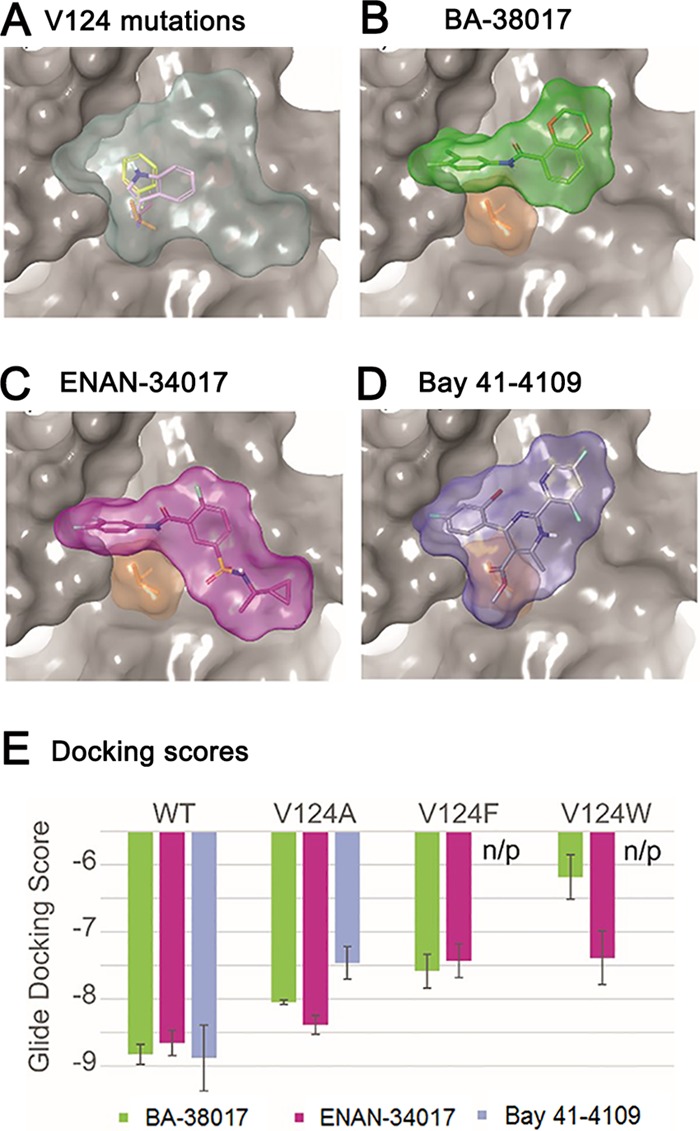
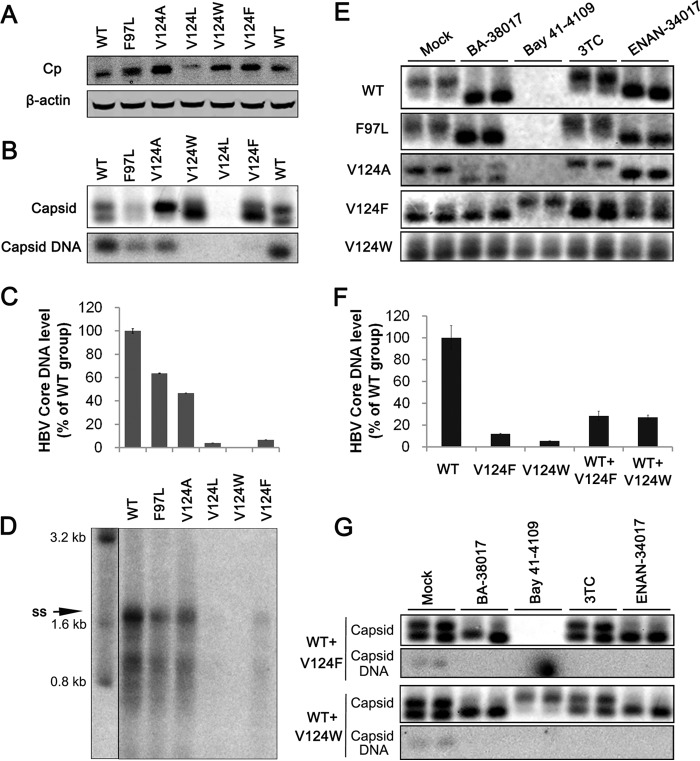
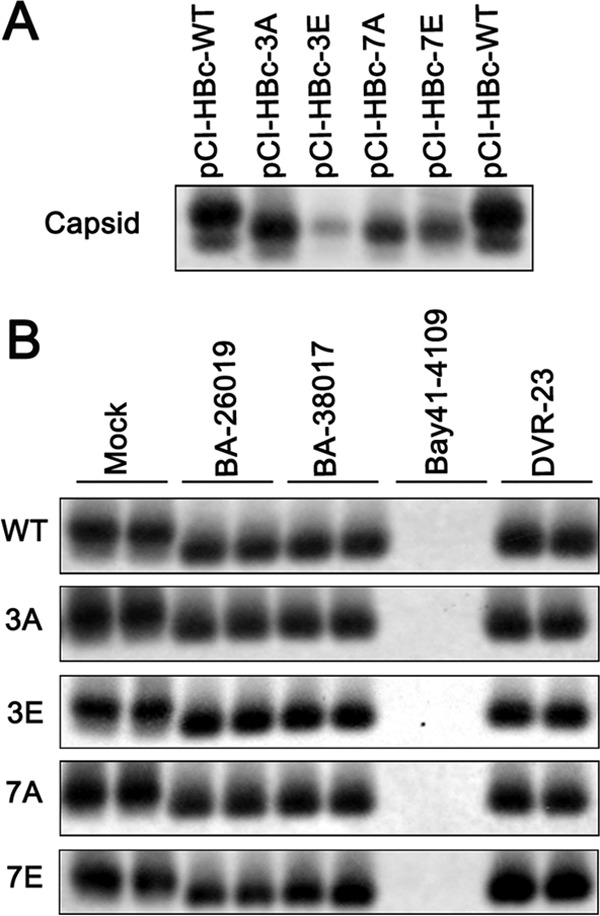
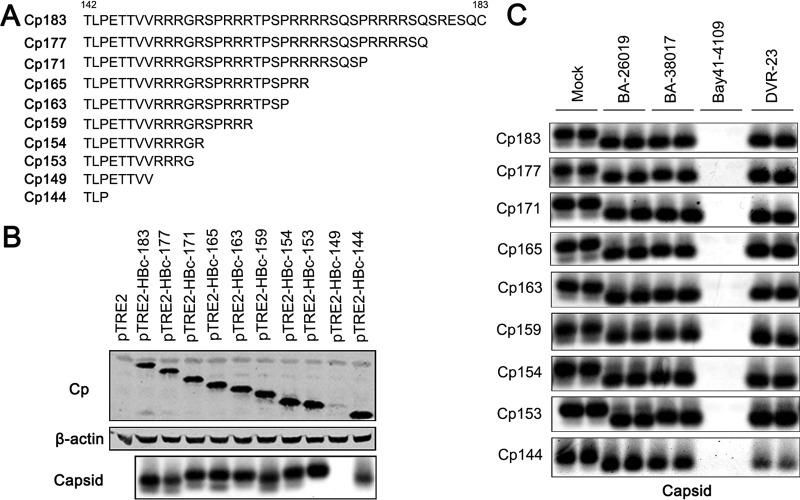
Chronic hepatitis B virus (HBV) infection is a global public health problem. Although the currently approved medications can reliably reduce the viral load and prevent the progression of liver diseases, they fail to cure the viral infection. In an effort toward discovery of novel antiviral agents against HBV, a group of benzamide (BA) derivatives that significantly reduced the amount of cytoplasmic HBV DNA were discovered. The initial lead optimization efforts identified two BA derivatives with improved antiviral activity for further mechanistic studies. Interestingly, similar to our previously reported sulfamoylbenzamides (SBAs), the BAs promote the formation of empty capsids through specific interaction with HBV core protein but not other viral and host cellular components. Genetic evidence suggested that both SBAs and BAs inhibited HBV nucleocapsid assembly by binding to the heteroaryldihydropyrimidine (HAP) pocket between core protein dimer-dimer interfaces. However, unlike SBAs, BA compounds uniquely induced the formation of empty capsids that migrated more slowly in native agarose gel electrophoresis from A36V mutant than from the wild-type core protein. Moreover, we showed that the assembly of chimeric capsids from wild-type and drug-resistant core proteins was susceptible to multiple capsid assembly modulators. Hence, HBV core protein is a dominant antiviral target that may suppress the selection of drug-resistant viruses during core protein-targeting antiviral therapy. Our studies thus indicate that BAs are a chemically and mechanistically unique type of HBV capsid assembly modulators and warranted for further development as antiviral agents against HBV.IMPORTANCE HBV core protein plays essential roles in many steps of the viral replication cycle. In addition to packaging viral pregenomic RNA (pgRNA) and DNA polymerase complex into nucleocapsids for reverse transcriptional DNA replication to take place, the core protein dimers, existing in several different quaternary structures in infected hepatocytes, participate in and regulate HBV virion assembly, capsid uncoating, and covalently closed circular DNA (cccDNA) formation. It is anticipated that small molecular core protein assembly modulators may disrupt one or multiple steps of HBV replication, depending on their interaction with the distinct quaternary structures of core protein. The discovery of novel core protein-targeting antivirals, such as benzamide derivatives reported here, and investigation of their antiviral mechanism may lead to the identification of antiviral therapeutics for the cure of chronic hepatitis B.
Keywords: antiviral agents; capsid assembly; hepatitis B virus.
Copyright © 2017 American Society for Microbiology.
Figures
References
-
- GBD 2013 Mortality and Causes of Death Collaborators. 2015. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385:117–171. doi:10.1016/S0140-6736(14)61682-2. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
