

The Number of Target Molecules of the Amplification Step Limits Accuracy and Sensitivity in Ultradeep-Sequencing Viral Population Studies
- PMID: 28566384
- PMCID: PMC5533907
- DOI: 10.1128/JVI.00561-17
The Number of Target Molecules of the Amplification Step Limits Accuracy and Sensitivity in Ultradeep-Sequencing Viral Population Studies
Abstract
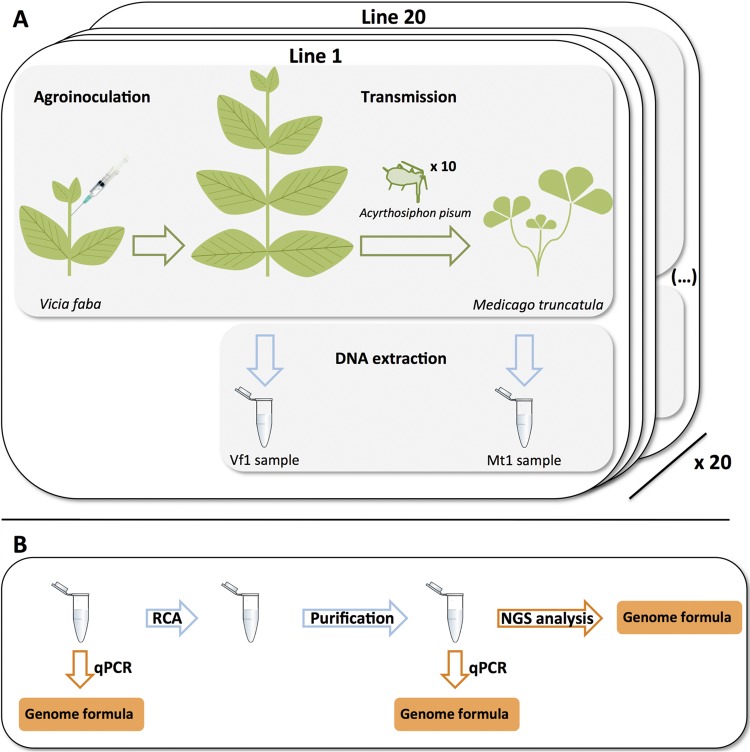
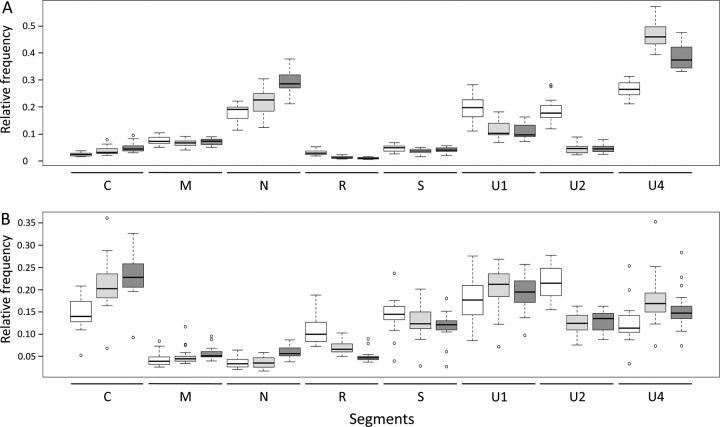
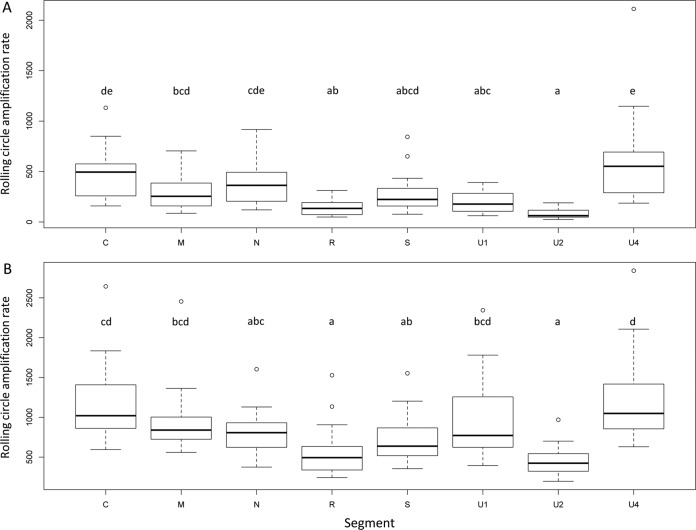
The invention of next-generation sequencing (NGS) techniques marked the coming of a new era in the detection of the genetic diversity of intrahost viral populations. A good understanding of the genetic structure of these populations requires, first, the ability to identify the different isolates or variants and, second, the ability to accurately quantify them. However, the initial amplification step of NGS studies can impose potential quantitative biases, modifying the variant relative frequencies. In particular, the number of target molecules (NTM) used during the amplification step is vastly overlooked although of primary importance, as it sets the limit of the accuracy and sensitivity of the sequencing procedure. In the present article, we investigated quantitative biases in an NGS study of populations of a multipartite single-stranded DNA (ssDNA) virus at different steps of the procedure. We studied 20 independent populations of the ssDNA virus faba bean necrotic stunt virus (FBNSV) in two host plants, Vicia faba and Medicago truncatula FBNSV is a multipartite virus composed of eight genomic segments, whose specific and host-dependent relative frequencies are defined as the "genome formula." Our results show a significant distortion of the FBNSV genome formula after the amplification and sequencing steps. We also quantified the genetic bottleneck occurring at the amplification step by documenting the NTM of two genomic segments of FBNSV. We argue that the NTM must be documented and carefully considered when determining the sensitivity and accuracy of data from NGS studies.IMPORTANCE The advent of next-generation sequencing (NGS) techniques now enables study of the genetic diversity of viral populations. A good understanding of the genetic structure of these populations first requires the ability to identify the different isolates or variants and second requires the ability to accurately quantify them. Prior to sequencing, viral genomes need to be amplified, a step that potentially imposes quantitative biases and modifies the viral population structure. In particular, the number of target molecules (NTM) used during the amplification step is of primary importance, as it sets the limit of the accuracy and sensitivity of the sequencing procedure. In this work, we used 20 replicated populations of the multipartite faba bean necrotic stunt virus (FBNSV) to estimate the various limitations of ultradeep-sequencing studies performed on intrahost viral populations. We report quantitative biases during rolling-circle amplification and the NTM of two genomic segments of FBNSV.
Keywords: DNA sequencing; FBNSV; Medicago truncatula; faba bean; next-generation sequencing; number of target molecules; rolling-circle amplification; sequencing accuracy; sequencing sensitivity.
Copyright © 2017 American Society for Microbiology.
Figures

Similar articles
-
Small Bottleneck Size in a Highly Multipartite Virus during a Complete Infection Cycle.J Virol. 2018 Jun 29;92(14):e00139-18. doi: 10.1128/JVI.00139-18. Print 2018 Jul 15. J Virol. 2018. PMID: 29720515 Free PMC article.
-
Replication-independent change in the frequencies of distinct genome segments of a multipartite virus during its transit within aphid vectors.Microbiol Spectr. 2024 May 2;12(5):e0028724. doi: 10.1128/spectrum.00287-24. Epub 2024 Mar 22. Microbiol Spectr. 2024. PMID: 38517168 Free PMC article.
-
First Report of a Nanovirus Disease of Pea in Germany.Plant Dis. 2010 May;94(5):642. doi: 10.1094/PDIS-94-5-0642C. Plant Dis. 2010. PMID: 30754436
-
Next-generation sequencing in clinical virology: Discovery of new viruses.World J Virol. 2015 Aug 12;4(3):265-76. doi: 10.5501/wjv.v4.i3.265. World J Virol. 2015. PMID: 26279987 Free PMC article. Review.
-
From Conventional to Next Generation Sequencing of Epstein-Barr Virus Genomes.Viruses. 2016 Feb 24;8(3):60. doi: 10.3390/v8030060. Viruses. 2016. PMID: 26927157 Free PMC article. Review.
Cited by
-
Barcoding of Plant Viruses with Circular Single-Stranded DNA Based on Rolling Circle Amplification.Viruses. 2018 Aug 31;10(9):469. doi: 10.3390/v10090469. Viruses. 2018. PMID: 30200312 Free PMC article. Review.
-
Nonconcomitant host-to-host transmission of multipartite virus genome segments may lead to complete genome reconstitution.Proc Natl Acad Sci U S A. 2022 Aug 9;119(32):e2201453119. doi: 10.1073/pnas.2201453119. Epub 2022 Aug 1. Proc Natl Acad Sci U S A. 2022. PMID: 35914138 Free PMC article.
-
A multicellular way of life for a multipartite virus.Elife. 2019 Mar 12;8:e43599. doi: 10.7554/eLife.43599. Elife. 2019. PMID: 30857590 Free PMC article.
-
Endemicity and prevalence of multipartite viruses under heterogeneous between-host transmission.PLoS Comput Biol. 2019 Mar 18;15(3):e1006876. doi: 10.1371/journal.pcbi.1006876. eCollection 2019 Mar. PLoS Comput Biol. 2019. PMID: 30883545 Free PMC article.
-
Co-Acquired Nanovirus and Geminivirus Exhibit a Contrasted Localization within Their Common Aphid Vector.Viruses. 2020 Mar 10;12(3):299. doi: 10.3390/v12030299. Viruses. 2020. PMID: 32164363 Free PMC article.
References
-
- Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, Robeson M, Edwards RA, Felts B, Rayhawk S, Knight R, Rohwer F, Jackson RB. 2007. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol 73:7059–7066. doi: 10.1128/AEM.00358-07. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
