

Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract
- PMID: 28566479
- PMCID: PMC5619971
- DOI: 10.1681/ASN.2017010043
Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract
Abstract
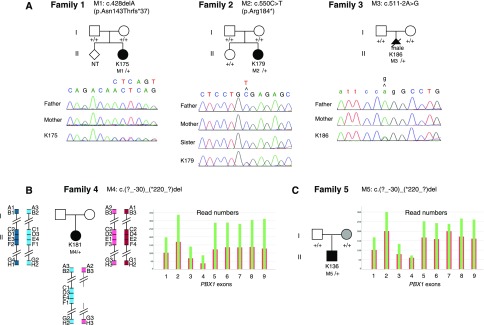
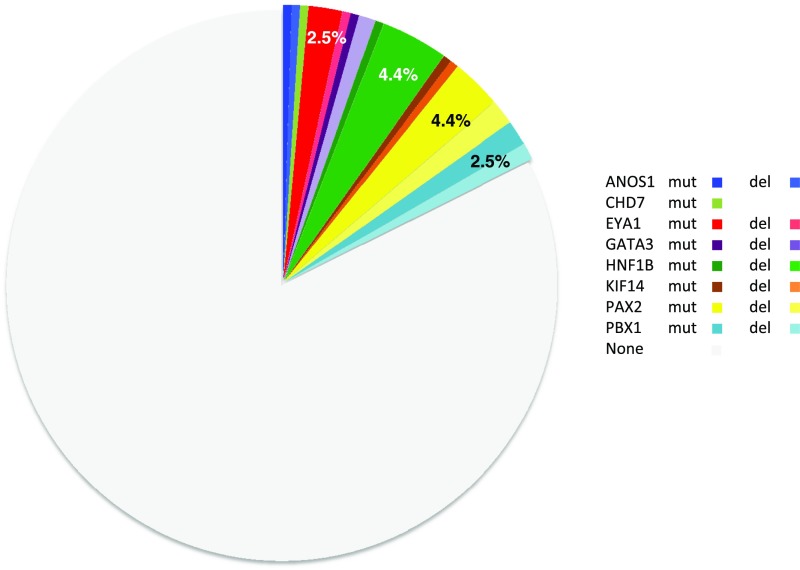
Congenital anomalies of the kidney and urinary tract (CAKUT) occur in three to six of 1000 live births, represent about 20% of the prenatally detected anomalies, and constitute the main cause of CKD in children. These disorders are phenotypically and genetically heterogeneous. Monogenic causes of CAKUT in humans and mice have been identified. However, despite high-throughput sequencing studies, the cause of the disease remains unknown in most patients, and several studies support more complex inheritance and the role of environmental factors and/or epigenetics in the pathophysiology of CAKUT. Here, we report the targeted exome sequencing of 330 genes, including genes known to be involved in CAKUT and candidate genes, in a cohort of 204 unrelated patients with CAKUT; 45% of the patients were severe fetal cases. We identified pathogenic mutations in 36 of 204 (17.6%) patients. These mutations included five de novo heterozygous loss of function mutations/deletions in the PBX homeobox 1 gene (PBX1), a gene known to have a crucial role in kidney development. In contrast, the frequency of SOX17 and DSTYK variants recently reported as pathogenic in CAKUT did not indicate causality. These findings suggest that PBX1 is involved in monogenic CAKUT in humans and call into question the role of some gene variants recently reported as pathogenic in CAKUT. Targeted exome sequencing also proved to be an efficient and cost-effective strategy to identify pathogenic mutations and deletions in known CAKUT genes.
Keywords: genetic renal disease; genetics and development; kidney development.
Copyright © 2017 by the American Society of Nephrology.
Figures
References
-
- Loane M, Dolk H, Kelly A, Teljeur C, Greenlees R, Densem J; EUROCAT Working Group : Paper 4: EUROCAT statistical monitoring: Identification and investigation of ten year trends of congenital anomalies in Europe. Birth Defects Res A Clin Mol Teratol 91[Suppl 1]: S31–S43, 2011 - PubMed
-
- Ardissino G, Daccò V, Testa S, Bonaudo R, Claris-Appiani A, Taioli E, Marra G, Edefonti A, Sereni F; ItalKid Project : Epidemiology of chronic renal failure in children: Data from the ItalKid project. Pediatrics 111: e382–e387, 2003 - PubMed
-
- Bulum B, Ozçakar ZB, Ustüner E, Düşünceli E, Kavaz A, Duman D, Walz K, Fitoz S, Tekin M, Yalçınkaya F: High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr Nephrol 28: 2143–2147, 2013 - PubMed
-
- Limwongse C: Syndromes and malformations of the urinary tract. In: Pediatric Nephrology, Vol. 1, 6th Ed., edited by Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Berlin, Springer, 2009, pp 121–156
-
- Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, Jankauskiené A, Mir S, Montini G, Peco-Antic A, Wühl E, Zurowska AM, Mehls O, Antignac C, Schaefer F, Salomon R: Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. J Am Soc Nephrol 17: 2864–2870, 2006 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
