

Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions
- PMID: 28572898
- PMCID: PMC5452277
- DOI: 10.1039/c6sc04157d
Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions
Abstract
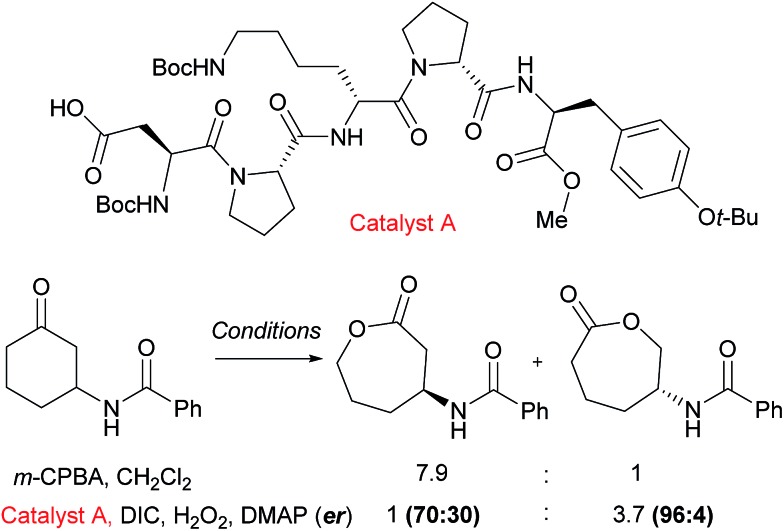
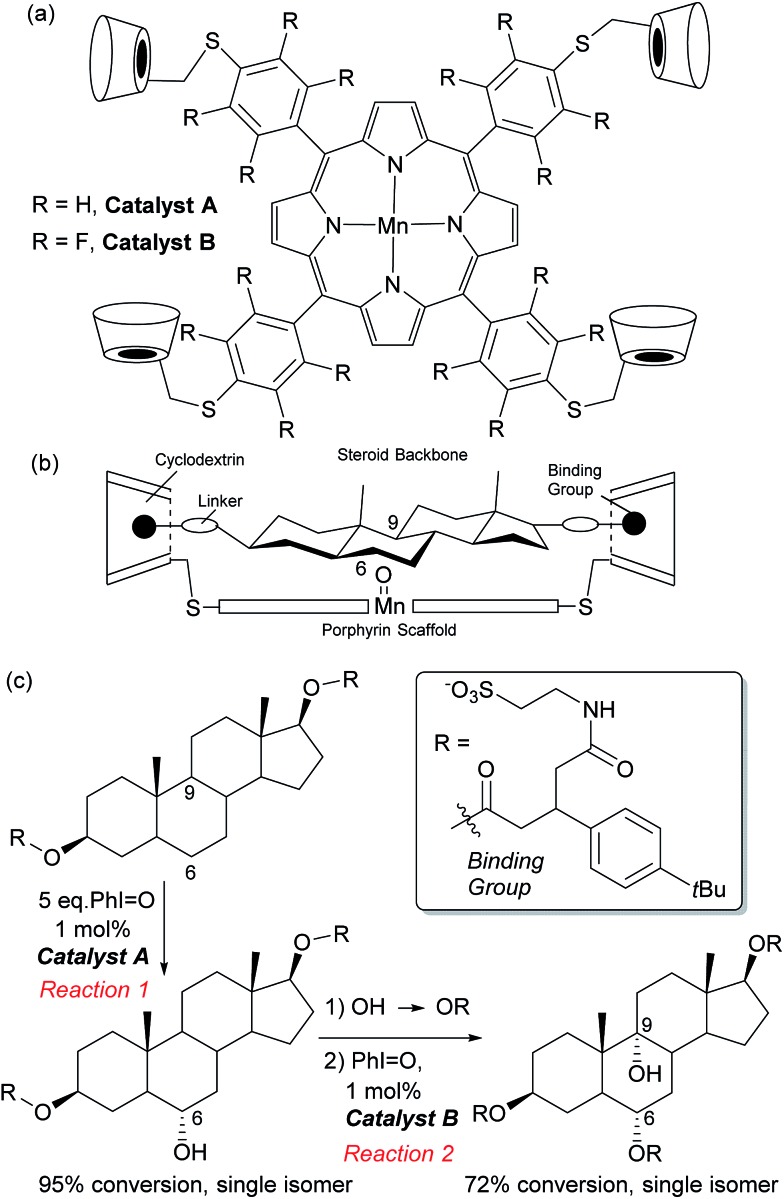
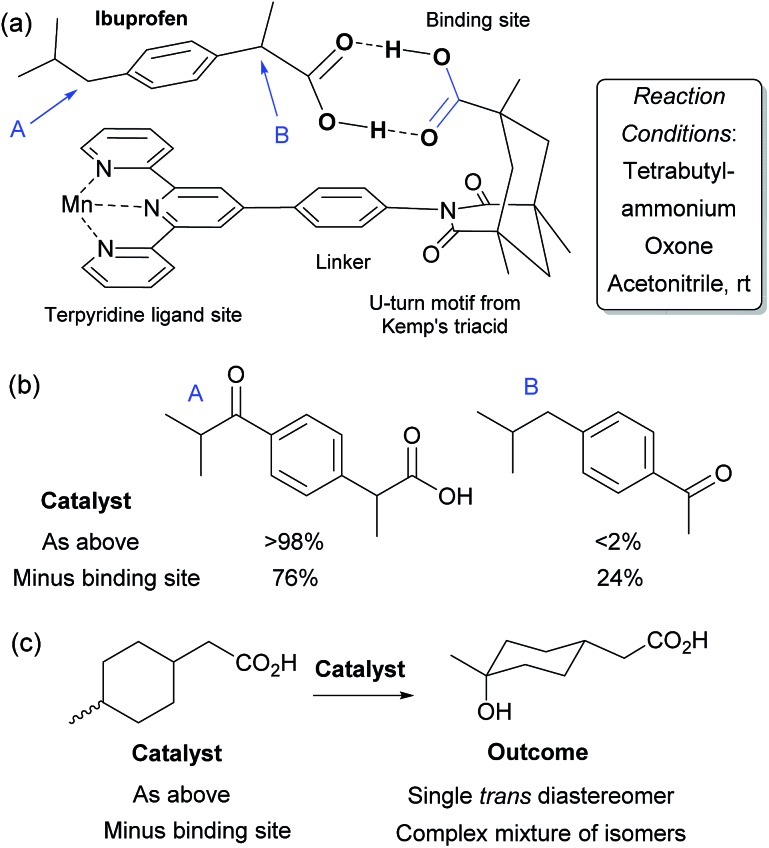
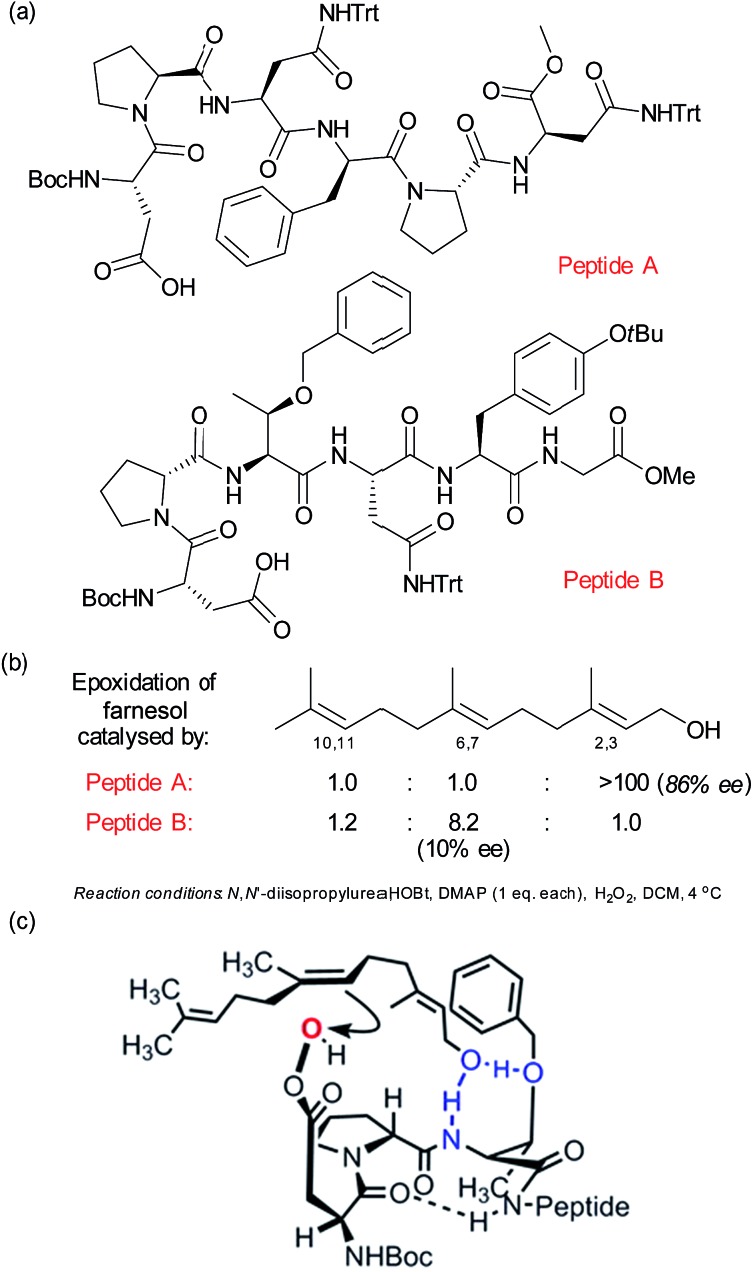
Asymmetric catalysis has been revolutionised by the realisation that attractive non-covalent interactions such as hydrogen bonds and ion pairs can act as powerful controllers of enantioselectivity when incorporated into appropriate small molecule chiral scaffolds. Given these tremendous advances it is surprising that there are still a relatively limited number of examples of non-covalent interactions being harnessed for control of regioselectivity or site-selectivity in catalysis, two other fundamental selectivity aspects facing the synthetic chemist. This perspective examines the progress that has been made in this area thus far using non-covalent interactions in conjunction with transition metal catalysis as well as in the context of purely organic catalysts. We hope this will highlight the great potential in this approach for designing selective catalytic reactions.
Figures
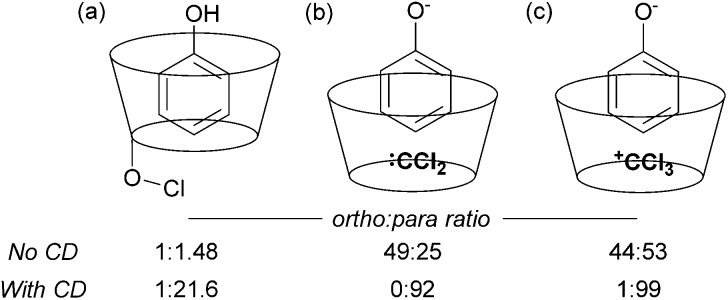
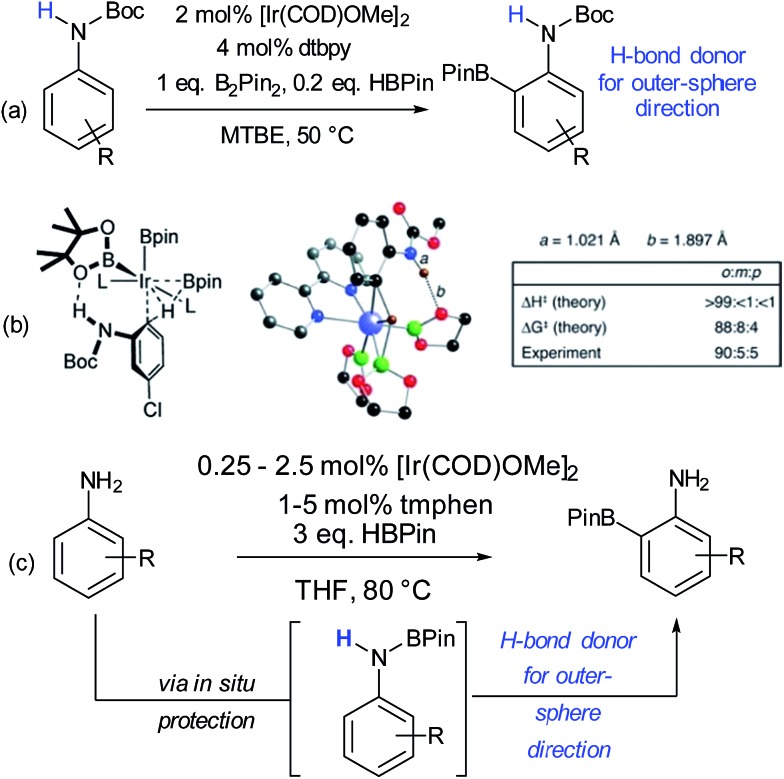
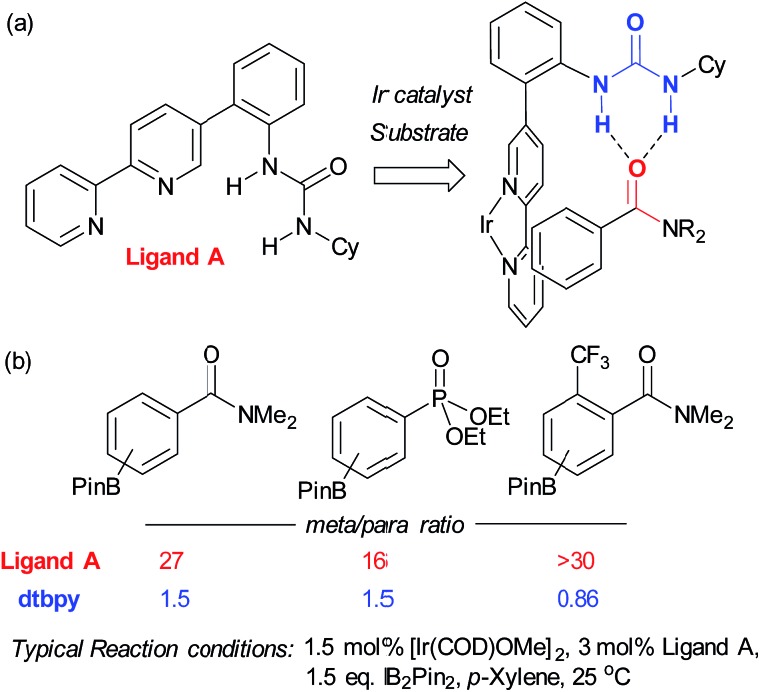
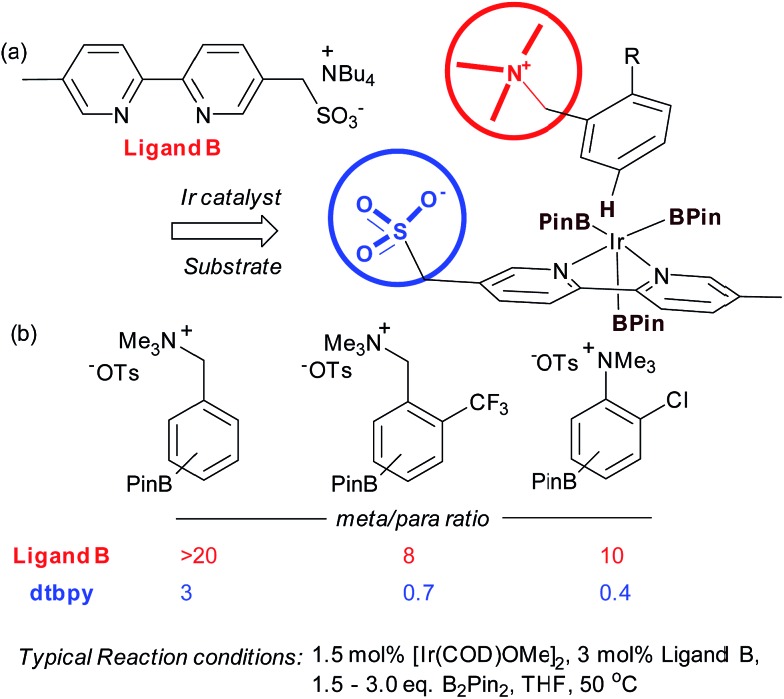
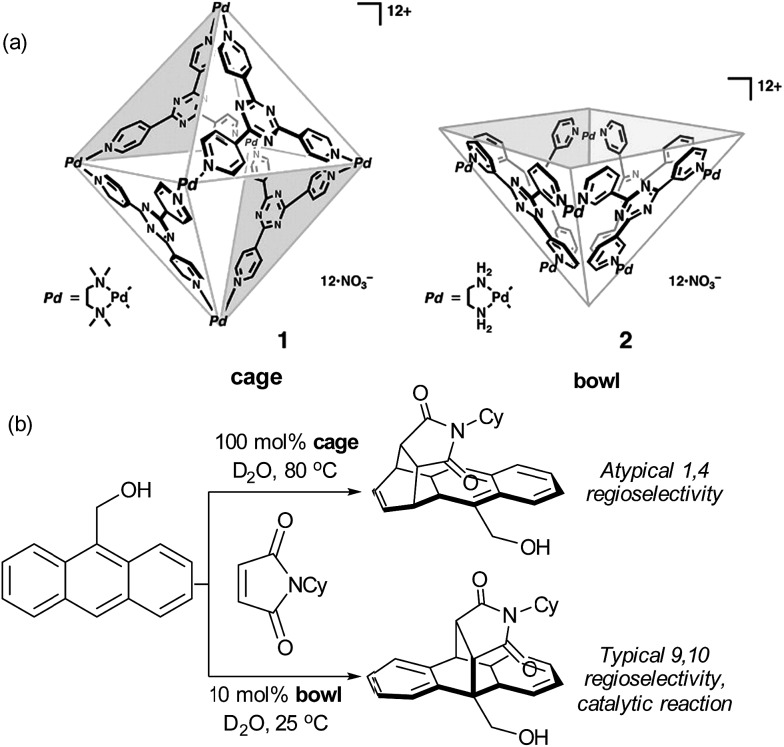
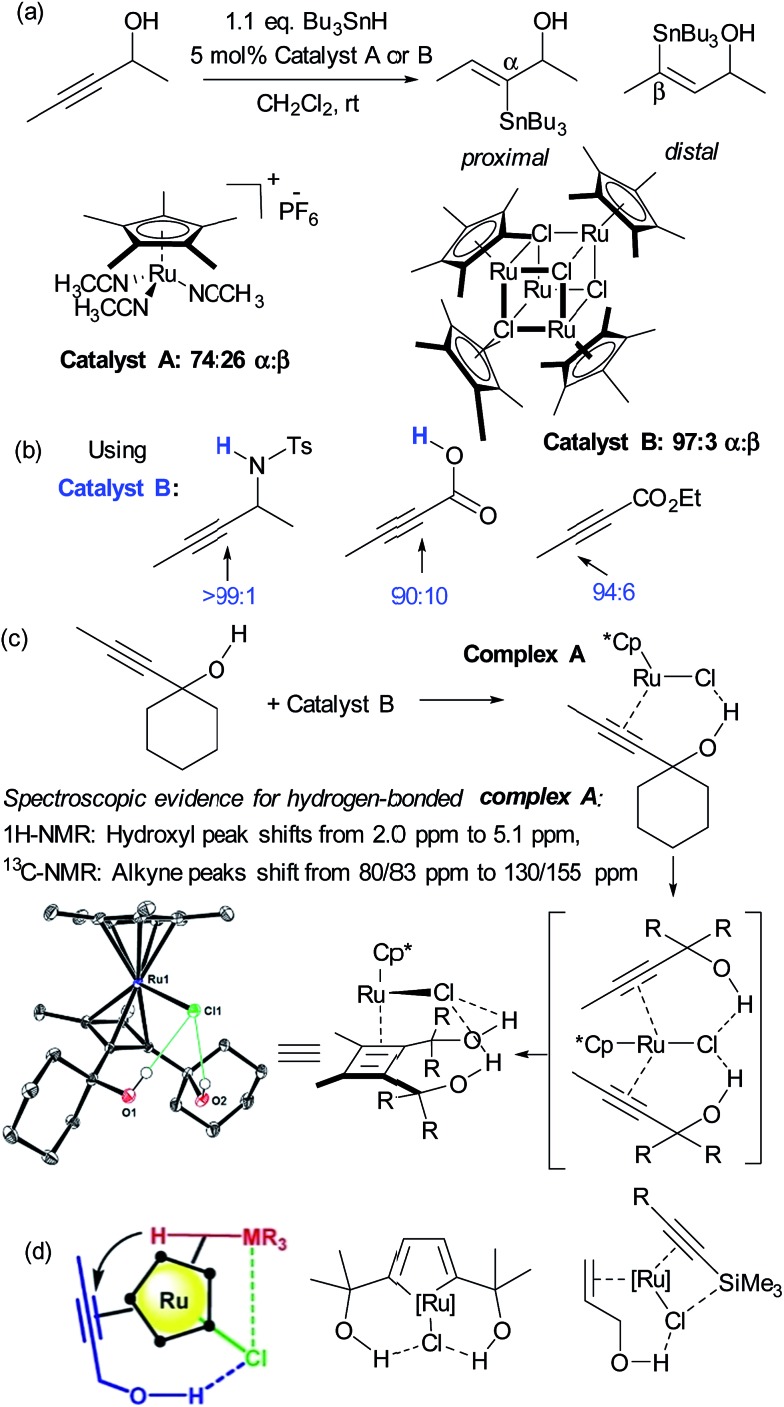
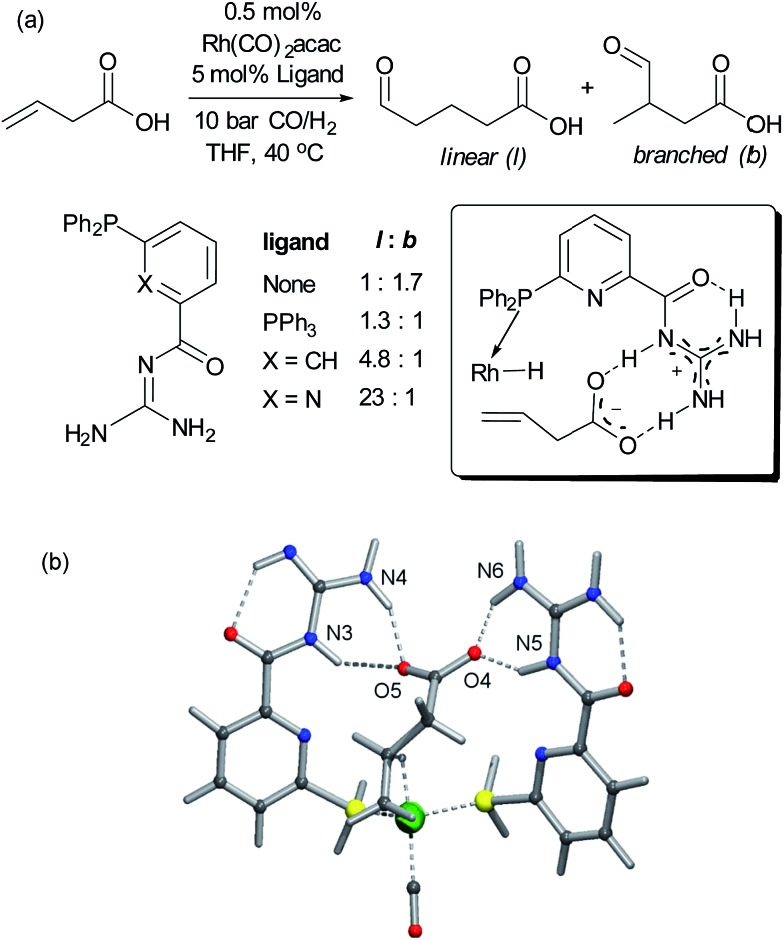
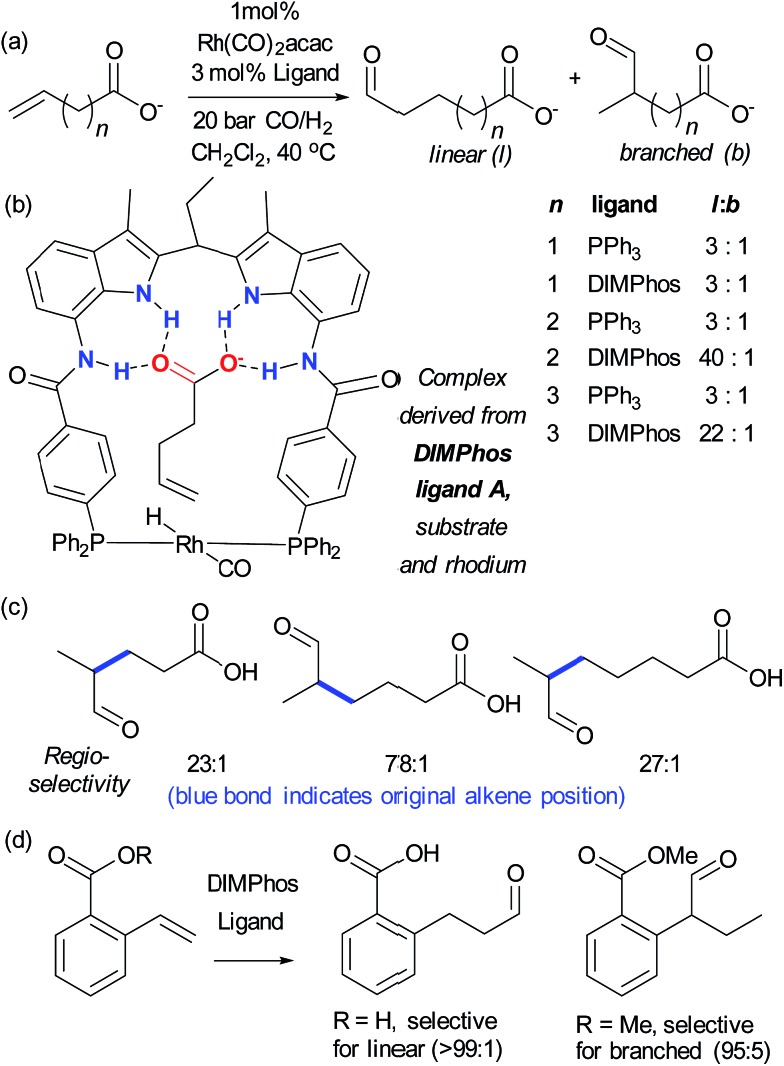
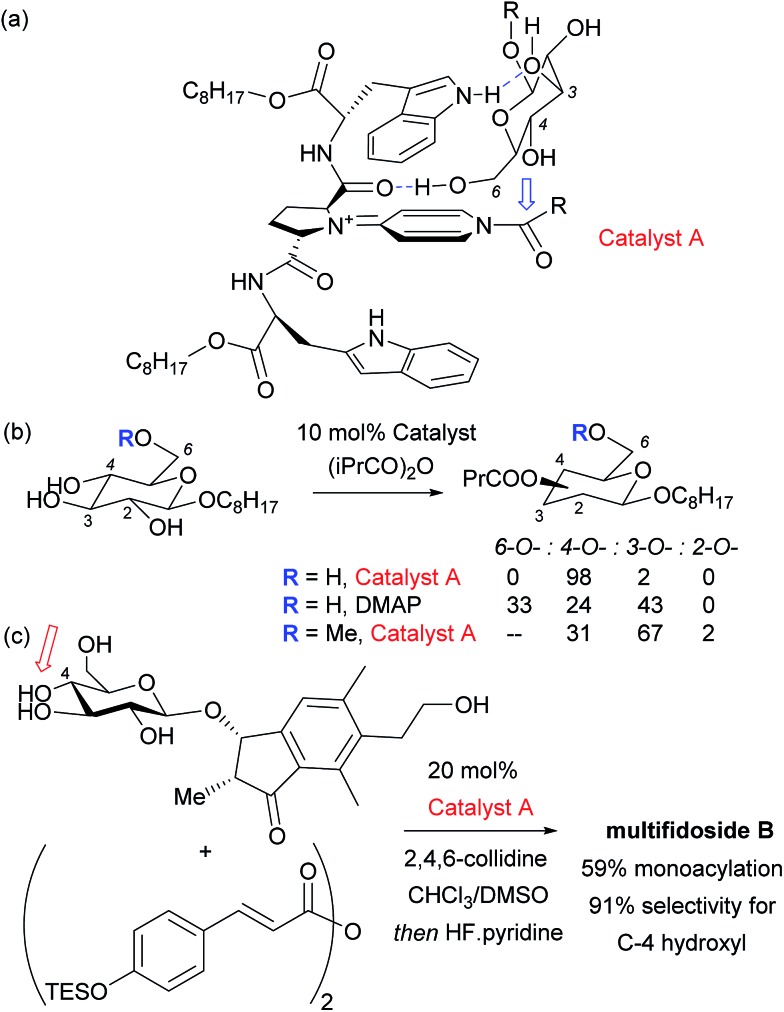
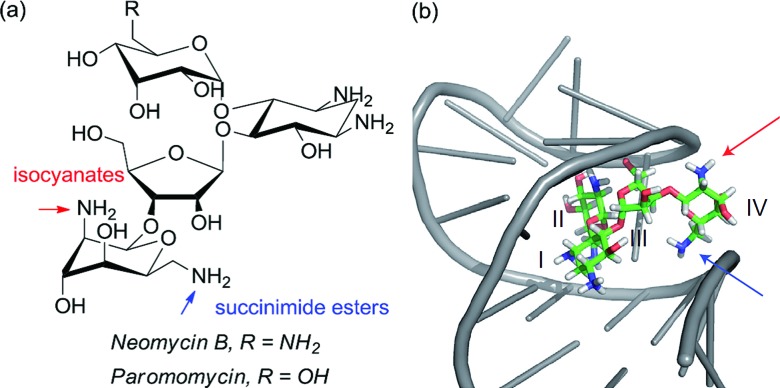
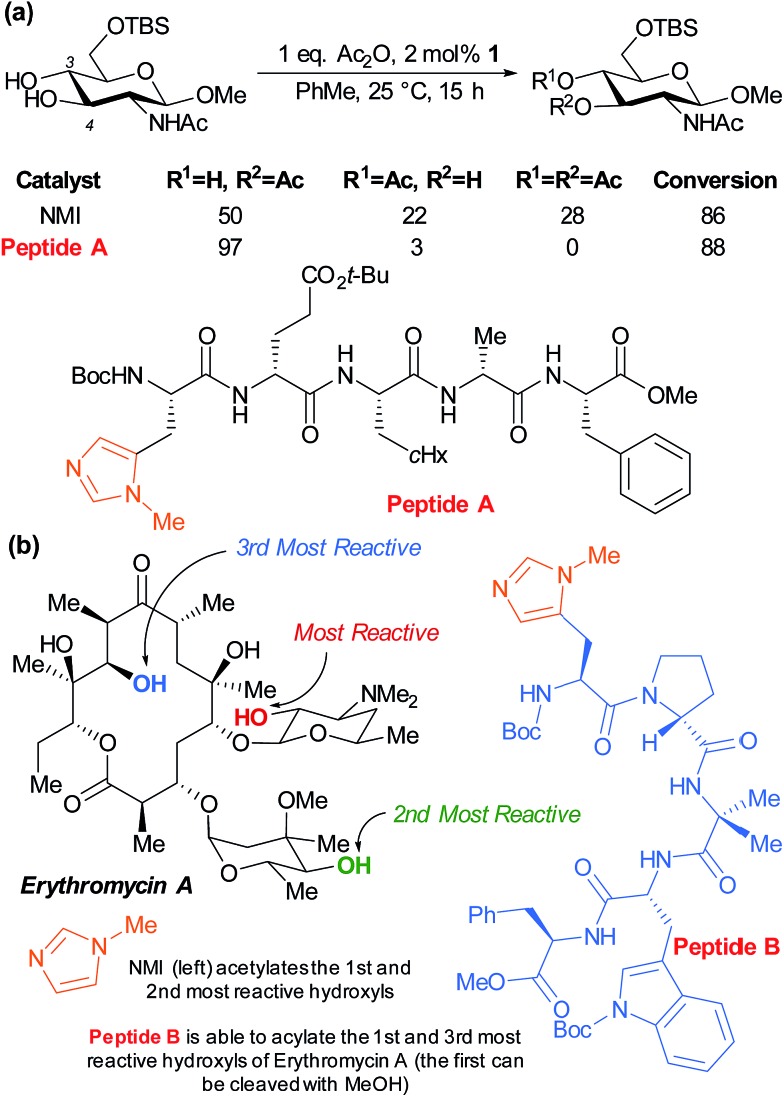
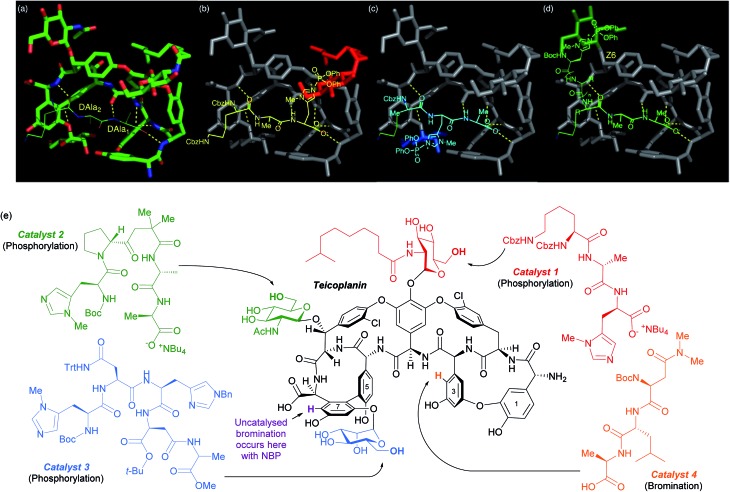


References
-
- Dalko P. I., Comprehensive Enantioselective Organocatalysis, Wiley, 2013.
-
- Pihko P. M., Hydrogen Bonding in Organic Synthesis, Wiley-VCH, Weinheim, 2009.
-
- Doyle A. G., Jacobsen E. N. Chem. Rev. 2007;107:5713–5743. - PubMed
-
- Turkmen Y., Zhu E. Y. and Rawal V. H., in Comprehensive Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley, Weiheim, 2013, vol. 1, ch. 12.
LinkOut - more resources
Full Text Sources
Other Literature Sources