

Loss of hepatic LRPPRC alters mitochondrial bioenergetics, regulation of permeability transition and trans-membrane ROS diffusion
- PMID: 28575497
- PMCID: PMC5886084
- DOI: 10.1093/hmg/ddx202
Loss of hepatic LRPPRC alters mitochondrial bioenergetics, regulation of permeability transition and trans-membrane ROS diffusion
Abstract
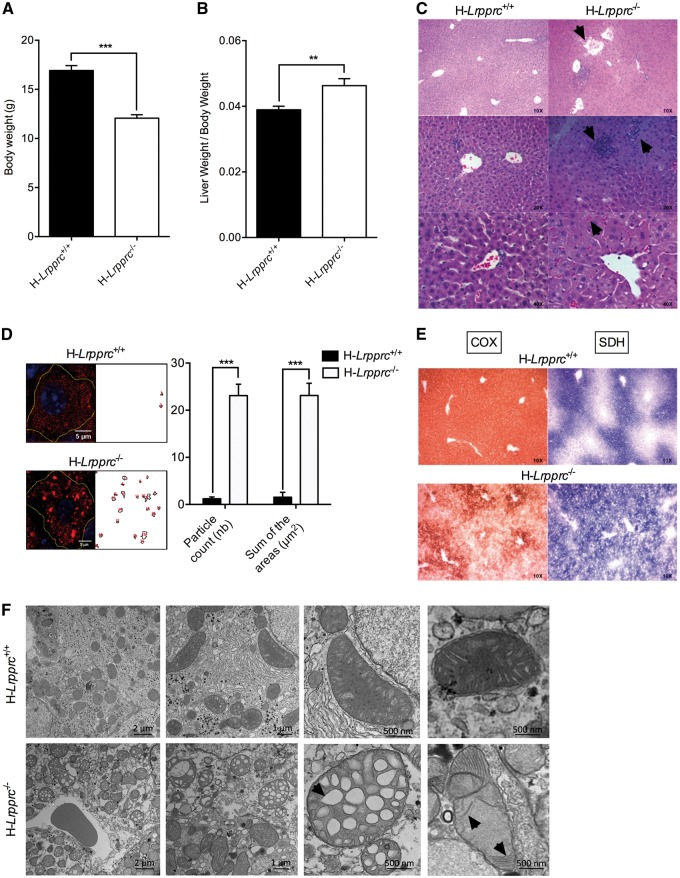
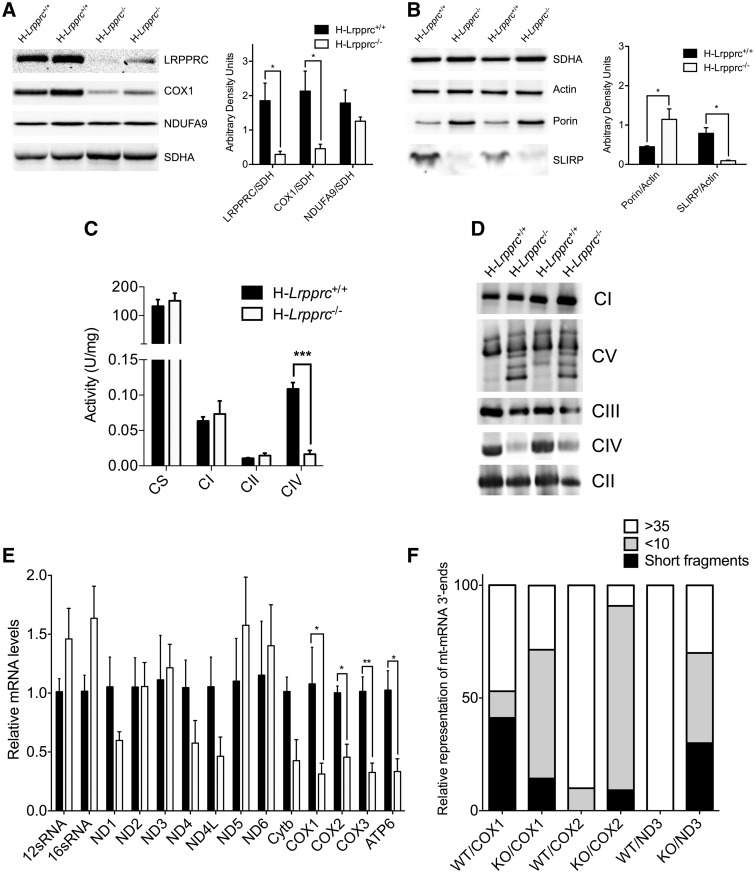
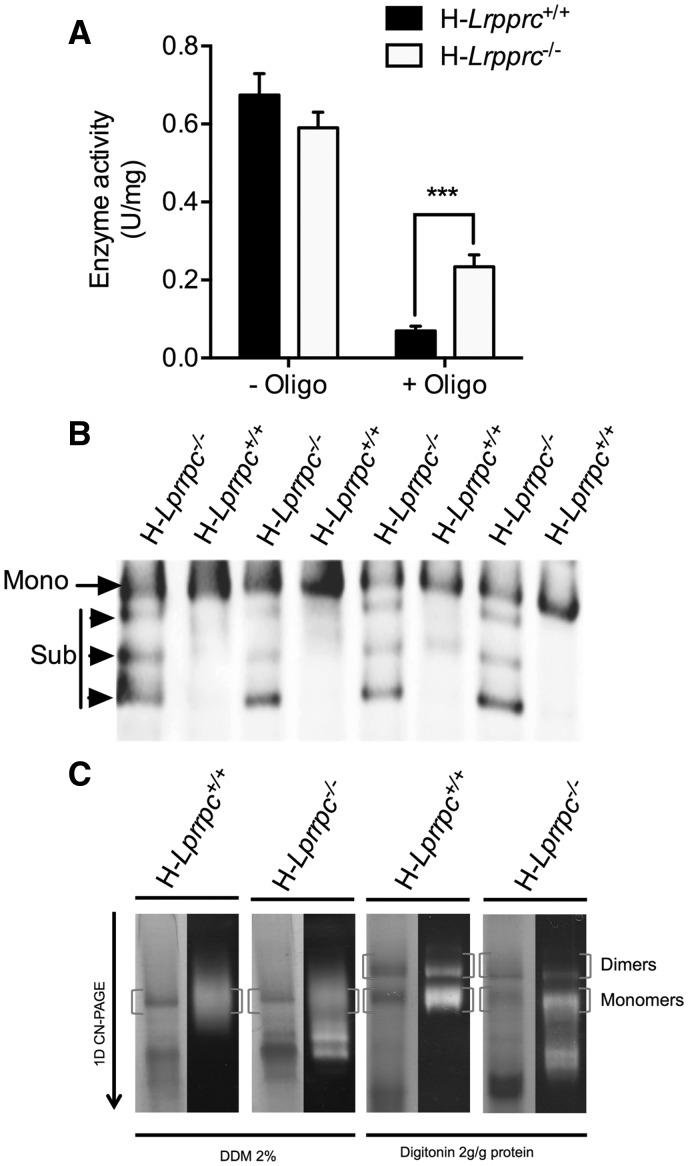
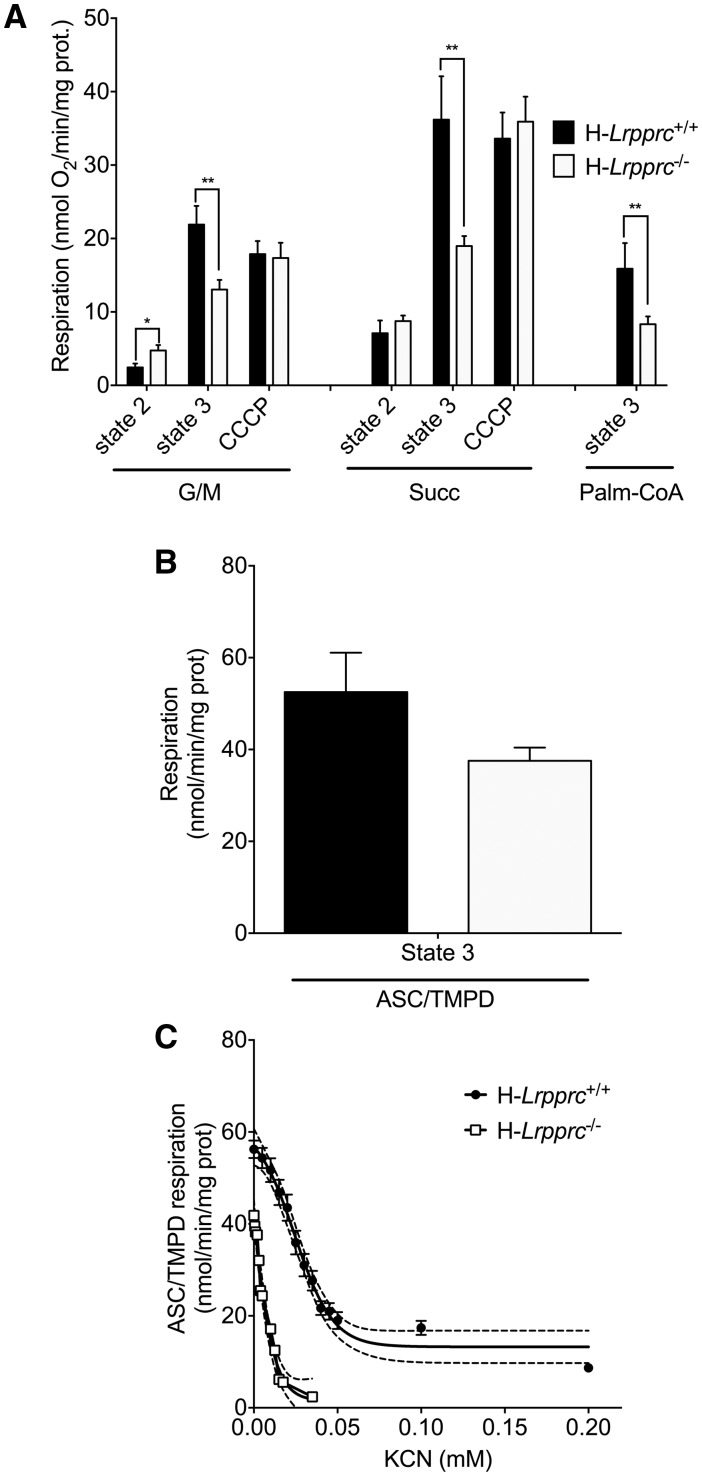
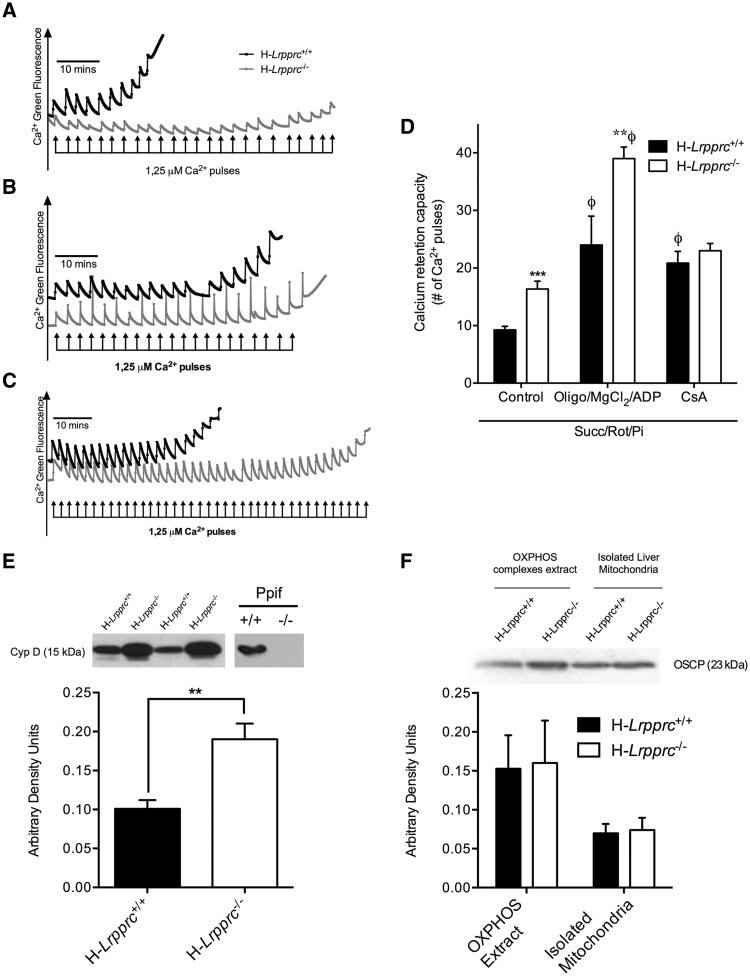
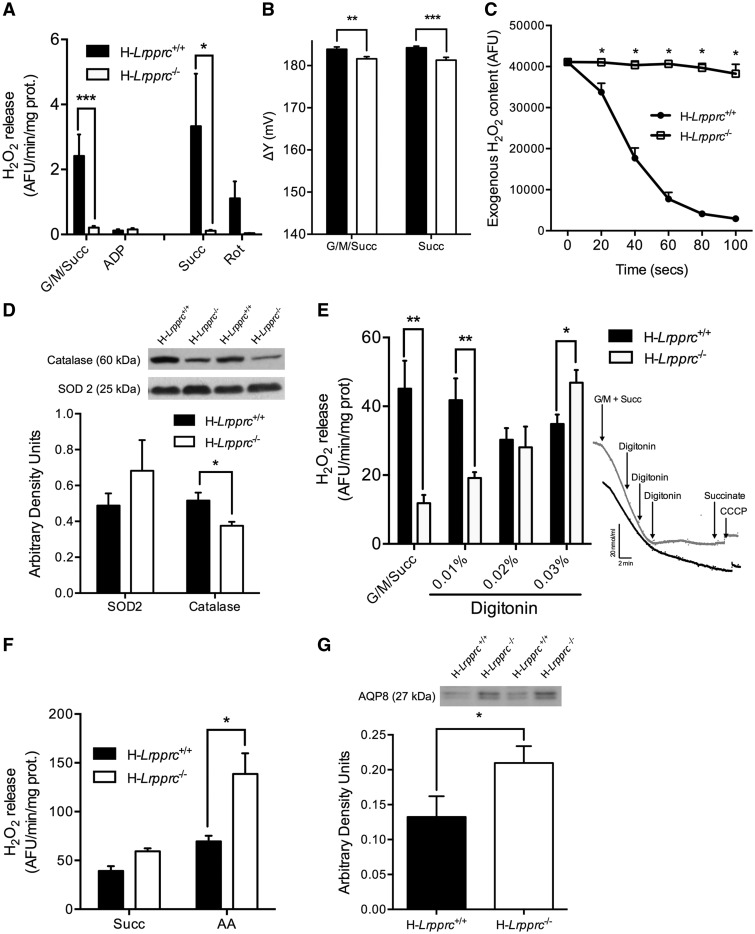
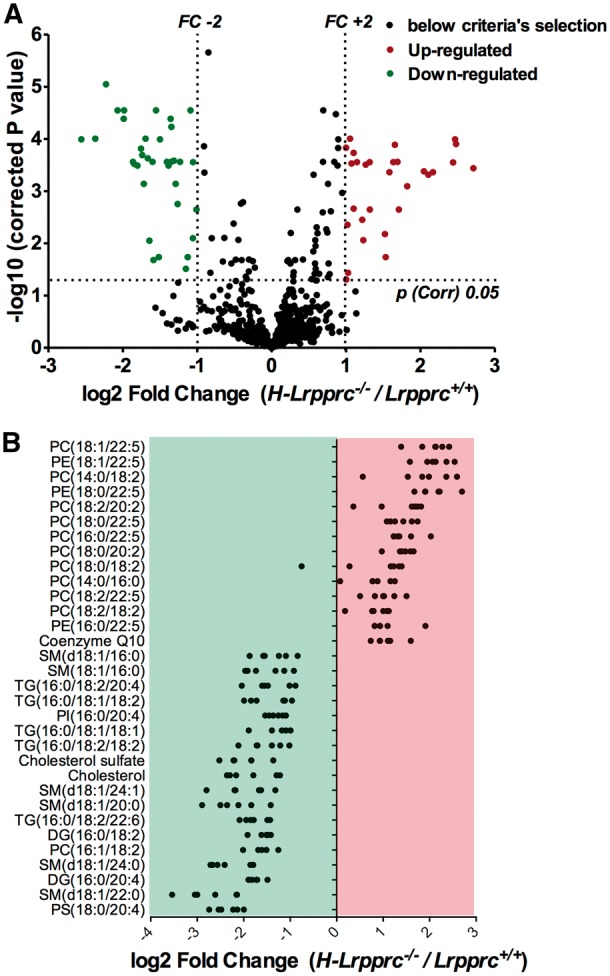
The French-Canadian variant of Leigh Syndrome (LSFC) is an autosomal recessive oxidative phosphorylation (OXPHOS) disorder caused by a mutation in LRPPRC, coding for a protein involved in the stability of mitochondrially-encoded mRNAs. Low levels of LRPPRC are present in all patient tissues, but result in a disproportionately severe OXPHOS defect in the brain and liver, leading to unpredictable subacute metabolic crises. To investigate the impact of the OXPHOS defect in the liver, we analyzed the mitochondrial phenotype in mice harboring an hepatocyte-specific inactivation of Lrpprc. Loss of LRPPRC in the liver caused a generalized growth delay, and typical histological features of mitochondrial hepatopathy. At the molecular level, LRPPRC deficiency caused destabilization of polyadenylated mitochondrial mRNAs, altered mitochondrial ultrastructure, and a severe complex IV (CIV) and ATP synthase (CV) assembly defect. The impact of LRPPRC deficiency was not limited to OXPHOS, but also included impairment of long-chain fatty acid oxidation, a striking dysregulation of the mitochondrial permeability transition pore, and an unsuspected alteration of trans-membrane H2O2 diffusion, which was traced to the ATP synthase assembly defect, and to changes in the lipid composition of mitochondrial membranes. This study underscores the value of mitochondria phenotyping to uncover complex and unexpected mechanisms contributing to the pathophysiology of mitochondrial disorders.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Debray F.-G., Lambert M., Mitchell G.A. (2008) Disorders of mitochondrial function. Curr. Opin. Pediatr., 20, 471–482. - PubMed
-
- Dimauro S., Schon E.A. (2008) Mitochondrial Disorders in the Nervous System. Annu. Rev. Neurosci., 31, 91–123. - PubMed
-
- Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M.. et al. (1998) SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet., 20, 337–343. - PubMed
-
- Rahman S., Blok R.B., Dahl H.H., Danks D.M., Kirby D.M., Chow C.W., Christodoulou J., Thorburn D.R. (1996) Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol., 39, 343–351. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
