

Mutations in SULT2B1 Cause Autosomal-Recessive Congenital Ichthyosis in Humans
- PMID: 28575648
- PMCID: PMC5473727
- DOI: 10.1016/j.ajhg.2017.05.007
Mutations in SULT2B1 Cause Autosomal-Recessive Congenital Ichthyosis in Humans
Abstract
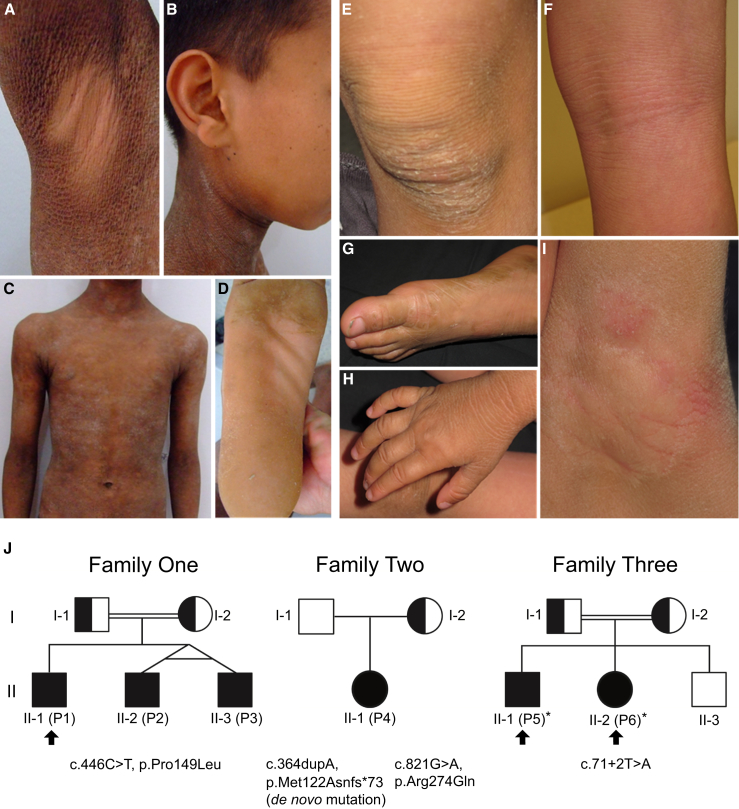
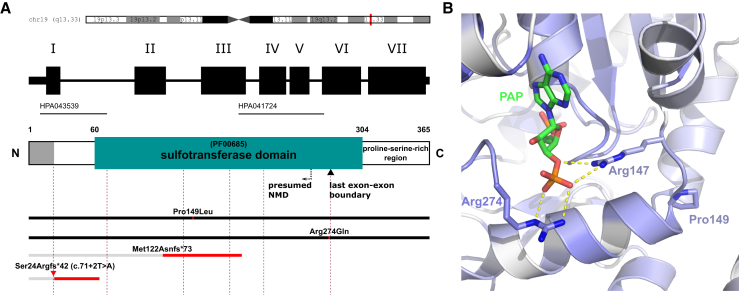
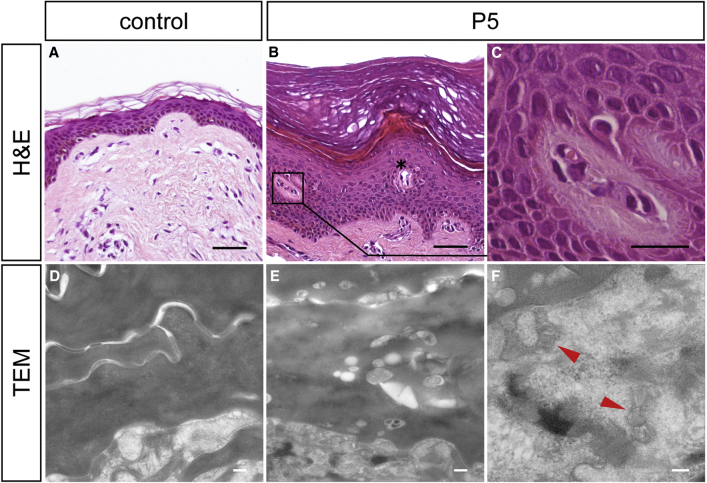
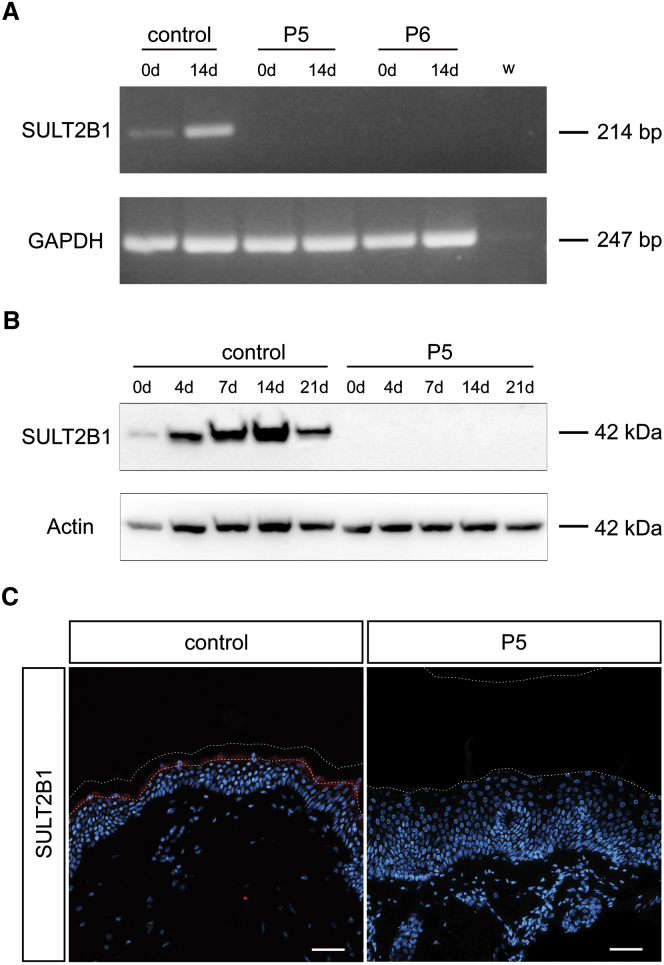
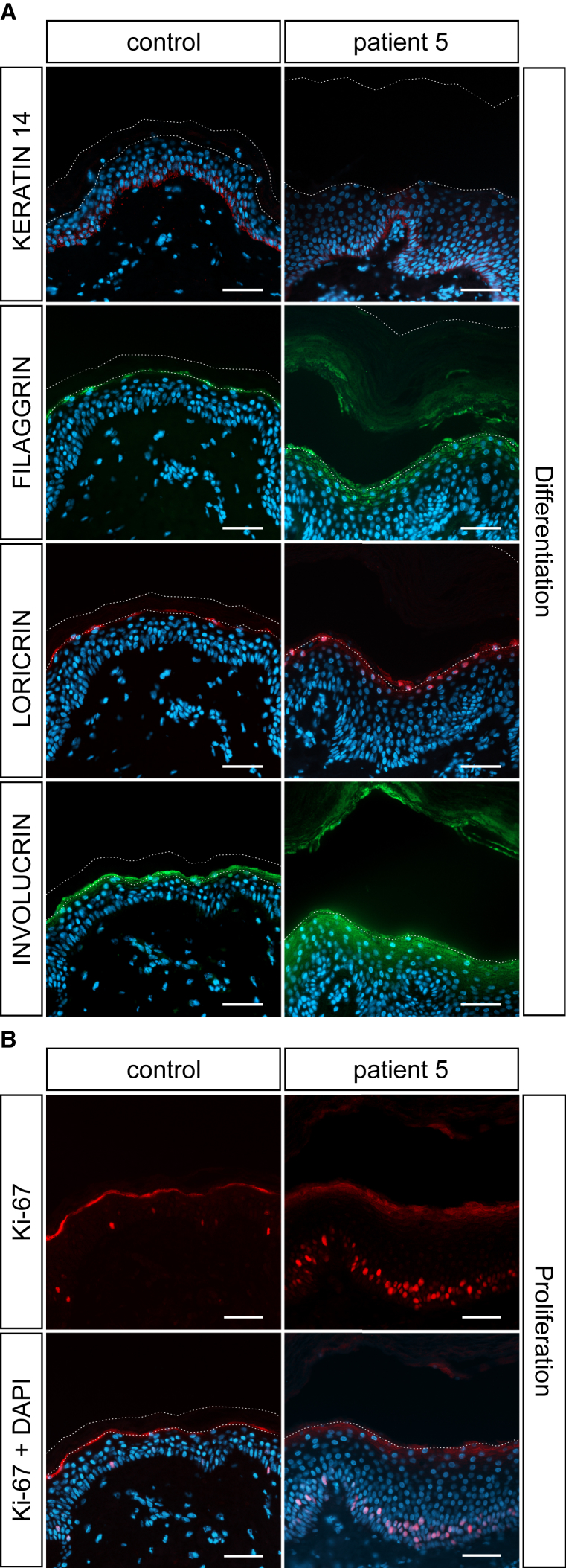
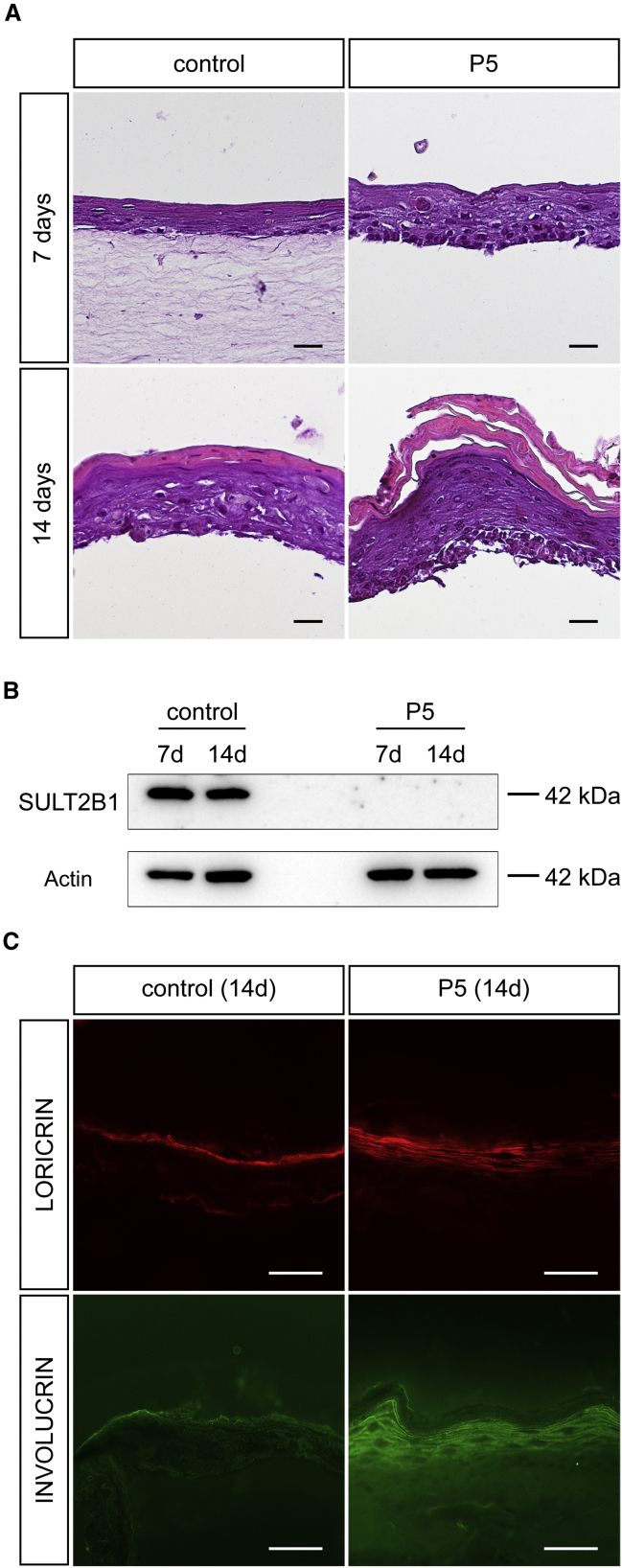
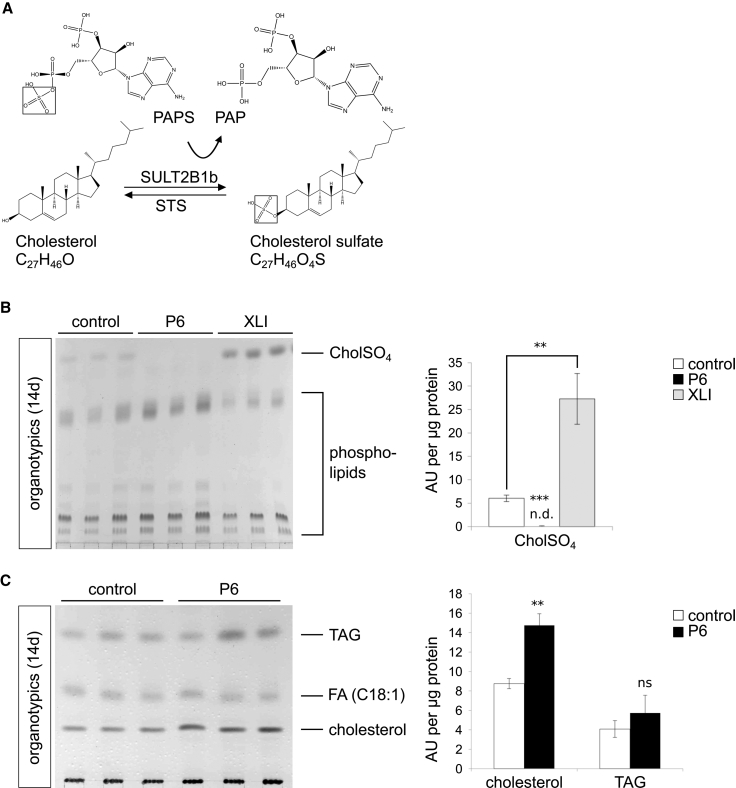
Ichthyoses are a clinically and genetically heterogeneous group of genodermatoses associated with abnormal scaling of the skin over the whole body. Mutations in nine genes are known to cause non-syndromic forms of autosomal-recessive congenital ichthyosis (ARCI). However, not all genetic causes for ARCI have been discovered to date. Using whole-exome sequencing (WES) and multigene panel screening, we identified 6 ARCI-affected individuals from three unrelated families with mutations in Sulfotransferase family 2B member 1 (SULT2B1), showing their causative association with ARCI. Cytosolic sulfotransferases form a large family of enzymes that are involved in the synthesis and metabolism of several steroids in humans. We identified four distinct mutations including missense, nonsense, and splice site mutations. We demonstrated the loss of SULT2B1 expression at RNA and protein levels in keratinocytes from individuals with ARCI by functional analyses. Furthermore, we succeeded in reconstructing the morphologic skin alterations in a 3D organotypic tissue culture model with SULT2B1-deficient keratinocytes and fibroblasts. By thin layer chromatography (TLC) of extracts from these organotypic cultures, we could show the absence of cholesterol sulfate, the metabolite of SULT2B1, and an increased level of cholesterol, indicating a disturbed cholesterol metabolism of the skin upon loss-of-function mutation in SULT2B1. In conclusion, our study reveals an essential role for SULT2B1 in the proper development of healthy human skin. Mutation in SULT2B1 leads to an ARCI phenotype via increased proliferation of human keratinocytes, thickening of epithelial layers, and altered epidermal cholesterol metabolism.
Keywords: 3D skin tissue culture model; SULT2B1; autosomal-recessive congenital ichthyosis; cholesterol sulfate cycle; epidermal differentiation; epidermal proliferation; epidermis; exome sequencing; ichthyosis; skin permeability barrier.
Copyright © 2017 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Oji V., Tadini G., Akiyama M., Blanchet Bardon C., Bodemer C., Bourrat E., Coudiere P., DiGiovanna J.J., Elias P., Fischer J. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 2010;63:607–641. - PubMed
-
- Traupe H., Fischer J., Oji V. Nonsyndromic types of ichthyoses - an update. J. Dtsch. Dermatol. Ges. 2014;12:109–121. - PubMed
-
- Huber M., Rettler I., Bernasconi K., Frenk E., Lavrijsen S.P., Ponec M., Bon A., Lautenschlager S., Schorderet D.F., Hohl D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995;267:525–528. - PubMed
-
- Russell L.J., DiGiovanna J.J., Rogers G.R., Steinert P.M., Hashem N., Compton J.G., Bale S.J. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat. Genet. 1995;9:279–283. - PubMed
-
- Jobard F., Lefèvre C., Karaduman A., Blanchet-Bardon C., Emre S., Weissenbach J., Ozgüc M., Lathrop M., Prud’homme J.-F., Fischer J. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum. Mol. Genet. 2002;11:107–113. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
