

Correlative super-resolution fluorescence and electron microscopy using conventional fluorescent proteins in vacuo
- PMID: 28576556
- PMCID: PMC5531056
- DOI: 10.1016/j.jsb.2017.05.013
Correlative super-resolution fluorescence and electron microscopy using conventional fluorescent proteins in vacuo
Abstract
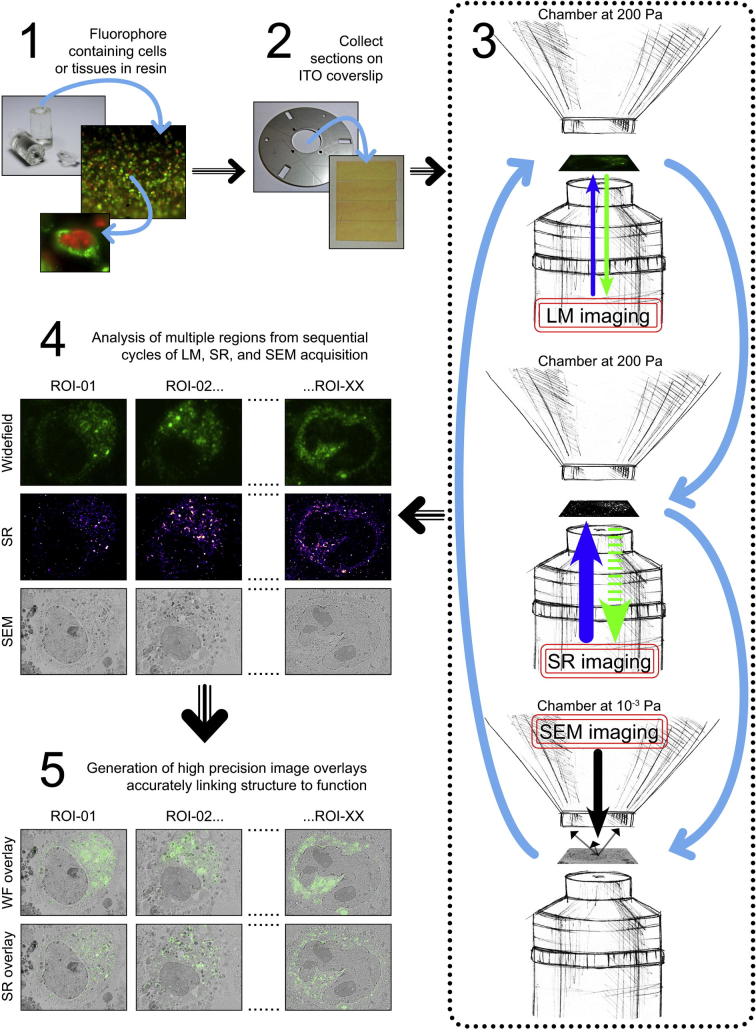
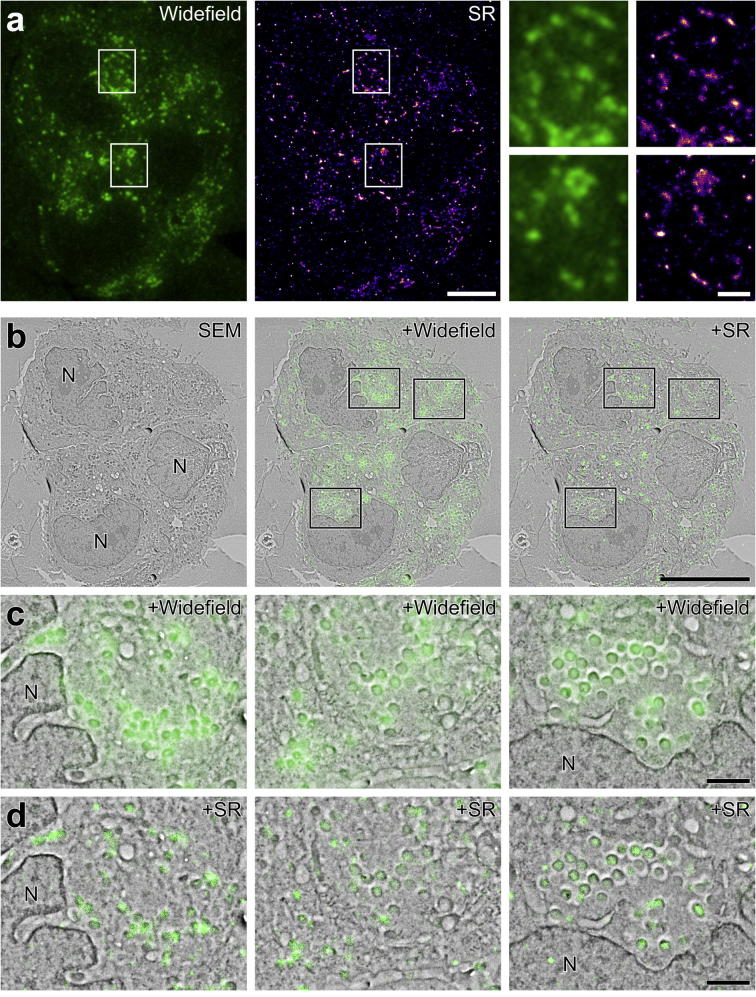
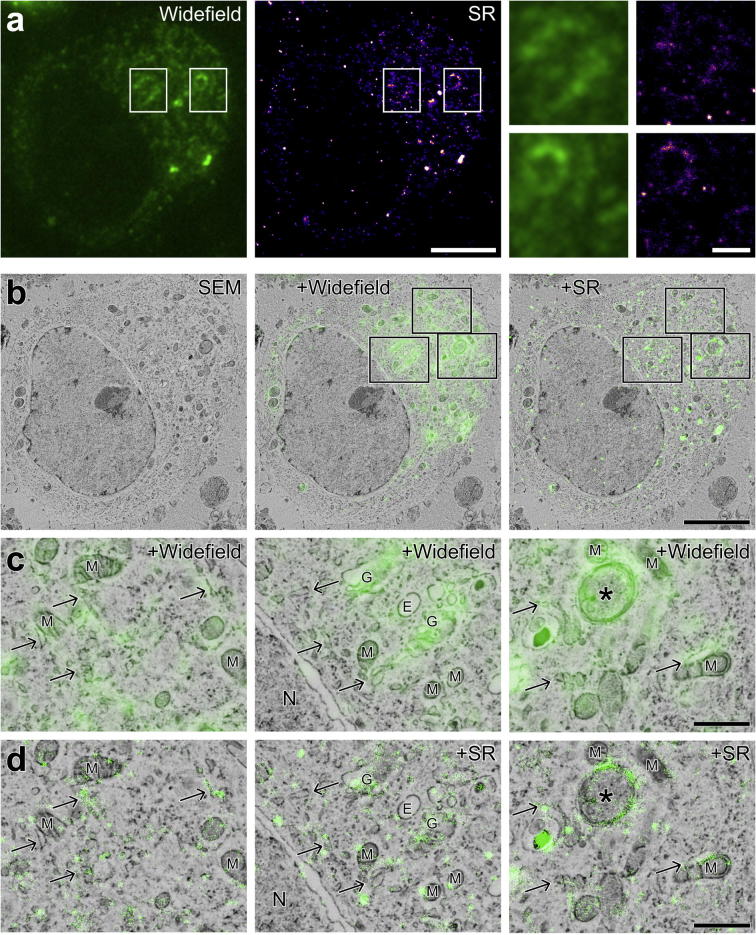
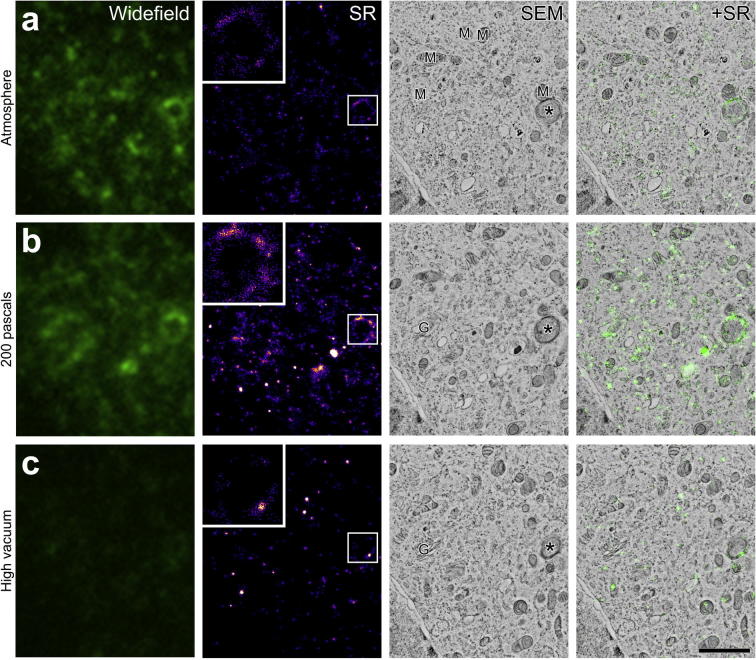
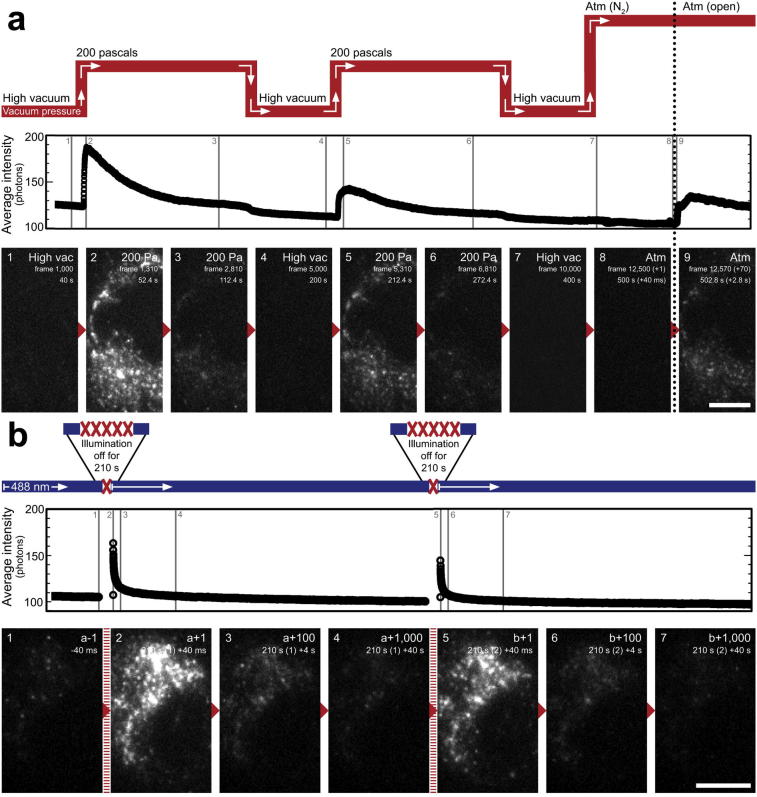
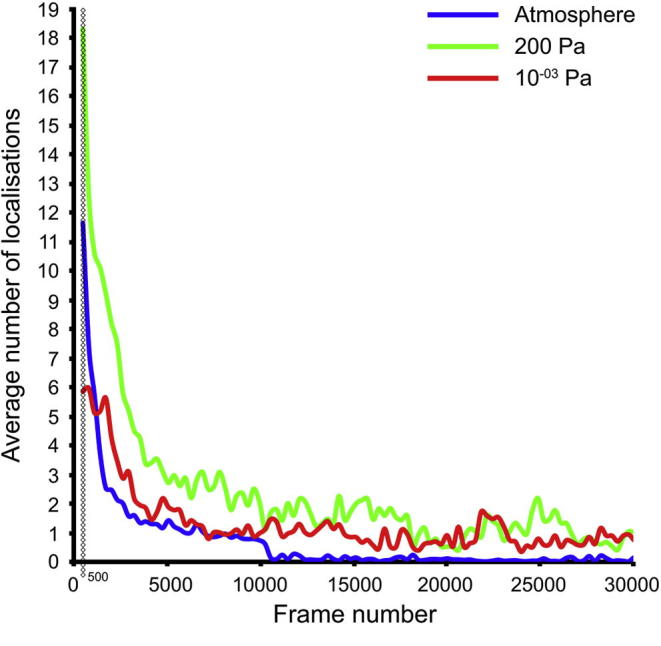
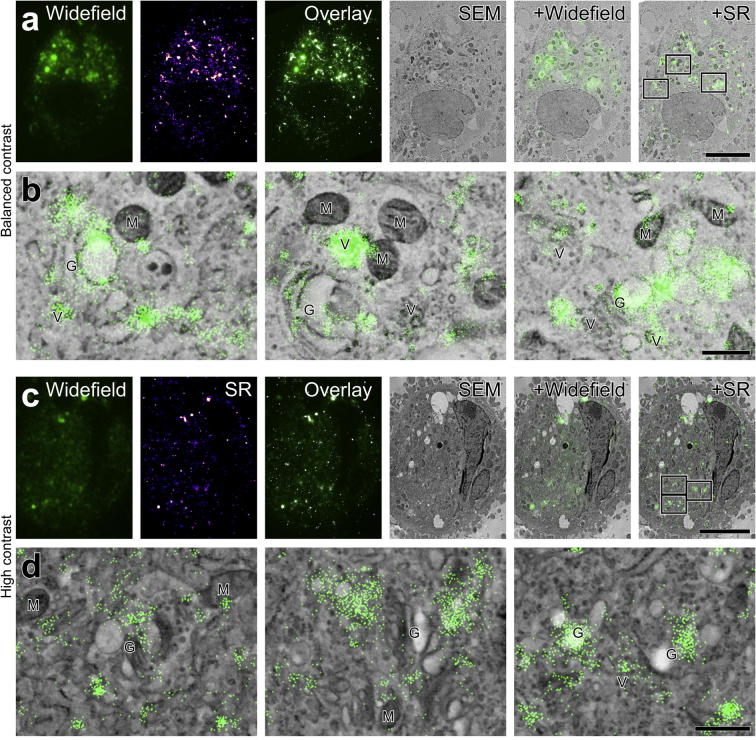
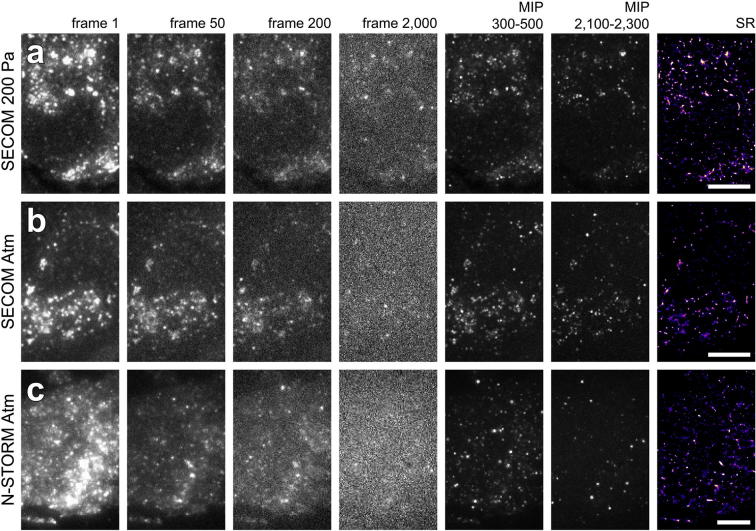
Super-resolution light microscopy, correlative light and electron microscopy, and volume electron microscopy are revolutionising the way in which biological samples are examined and understood. Here, we combine these approaches to deliver super-accurate correlation of fluorescent proteins to cellular structures. We show that YFP and GFP have enhanced blinking properties when embedded in acrylic resin and imaged under partial vacuum, enabling in vacuo single molecule localisation microscopy. In conventional section-based correlative microscopy experiments, the specimen must be moved between imaging systems and/or further manipulated for optimal viewing. These steps can introduce undesirable alterations in the specimen, and complicate correlation between imaging modalities. We avoided these issues by using a scanning electron microscope with integrated optical microscope to acquire both localisation and electron microscopy images, which could then be precisely correlated. Collecting data from ultrathin sections also improved the axial resolution and signal-to-noise ratio of the raw localisation microscopy data. Expanding data collection across an array of sections will allow 3-dimensional correlation over unprecedented volumes. The performance of this technique is demonstrated on vaccinia virus (with YFP) and diacylglycerol in cellular membranes (with GFP).
Keywords: 3-dimensional; Blinking; CLEM; Correlative; Electron microscopy; Fluorescence; GFP; ILSEM; In-resin fluorescence; Integrated; Protein localisation; Super-resolution; Volume; YFP.
Copyright © 2017. Published by Elsevier Inc.
Figures

Similar articles
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
-
Comparison of cellulose, modified cellulose and synthetic membranes in the haemodialysis of patients with end-stage renal disease.Cochrane Database Syst Rev. 2001;(3):CD003234. doi: 10.1002/14651858.CD003234. Cochrane Database Syst Rev. 2001. Update in: Cochrane Database Syst Rev. 2005 Jul 20;(3):CD003234. doi: 10.1002/14651858.CD003234.pub2. PMID: 11687058 Updated.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2017 Dec 22;12(12):CD011535. doi: 10.1002/14651858.CD011535.pub2. Cochrane Database Syst Rev. 2017. Update in: Cochrane Database Syst Rev. 2020 Jan 9;1:CD011535. doi: 10.1002/14651858.CD011535.pub3. PMID: 29271481 Free PMC article. Updated.
-
Magnetic resonance perfusion for differentiating low-grade from high-grade gliomas at first presentation.Cochrane Database Syst Rev. 2018 Jan 22;1(1):CD011551. doi: 10.1002/14651858.CD011551.pub2. Cochrane Database Syst Rev. 2018. PMID: 29357120 Free PMC article.
-
Diagnostic test accuracy and cost-effectiveness of tests for codeletion of chromosomal arms 1p and 19q in people with glioma.Cochrane Database Syst Rev. 2022 Mar 2;3(3):CD013387. doi: 10.1002/14651858.CD013387.pub2. Cochrane Database Syst Rev. 2022. PMID: 35233774 Free PMC article.
Cited by
-
Direct imaging of uncoated biological samples enables correlation of super-resolution and electron microscopy data.Sci Rep. 2018 Aug 2;8(1):11610. doi: 10.1038/s41598-018-29970-x. Sci Rep. 2018. PMID: 30072703 Free PMC article.
-
Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells.Science. 2020 Jan 17;367(6475):eaaz5357. doi: 10.1126/science.aaz5357. Science. 2020. PMID: 31949053 Free PMC article.
-
High-precision targeting workflow for volume electron microscopy.J Cell Biol. 2021 Sep 6;220(9):e202104069. doi: 10.1083/jcb.202104069. Epub 2021 Jun 23. J Cell Biol. 2021. PMID: 34160561 Free PMC article.
-
Visualization of cytoplasmic organelles via in-resin CLEM using an osmium-resistant far-red protein.Sci Rep. 2020 Jul 9;10(1):11314. doi: 10.1038/s41598-020-68191-z. Sci Rep. 2020. PMID: 32647231 Free PMC article.
-
Correlative light and electron microscopy reveals fork-shaped structures at actin entry sites of focal adhesions.Biol Open. 2022 Nov 1;11(11):bio059417. doi: 10.1242/bio.059417. Epub 2022 Nov 21. Biol Open. 2022. PMID: 36409550 Free PMC article.
References
-
- Arakawa Y., Cordeiro J.V., Schleich S., Newsome T.P., Way M. The release of vaccinia virus from infected cells requires RhoA-mDia modulation of cortical actin. Cell Host Microbe. 2007;1:227–240. - PubMed
-
- Bagshaw C.R., Cherny D. Blinking fluorophores: what do they tell us about protein dynamics? Biochem. Soc. Trans. 2006;34:979–982. - PubMed
-
- Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S., Bonifacino J.S., Davidson M.W., Lippincott-Schwartz J., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. - PubMed
-
- Brama E., Peddie C.J., Jones M.L., Domart M.-C., Snetkov X., Way M., Larijani B., Collinson L.M. Standard fluorescent proteins as dual-modality probes for correlative experiments in an integrated light and electron microscope. J. Chem. Biol. 2015;8:179–188.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
