

Modeling gene regulation from paired expression and chromatin accessibility data
- PMID: 28576882
- PMCID: PMC5488952
- DOI: 10.1073/pnas.1704553114
Modeling gene regulation from paired expression and chromatin accessibility data
Abstract
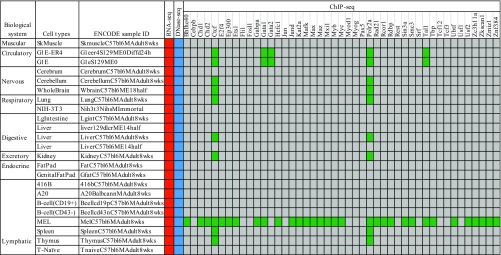
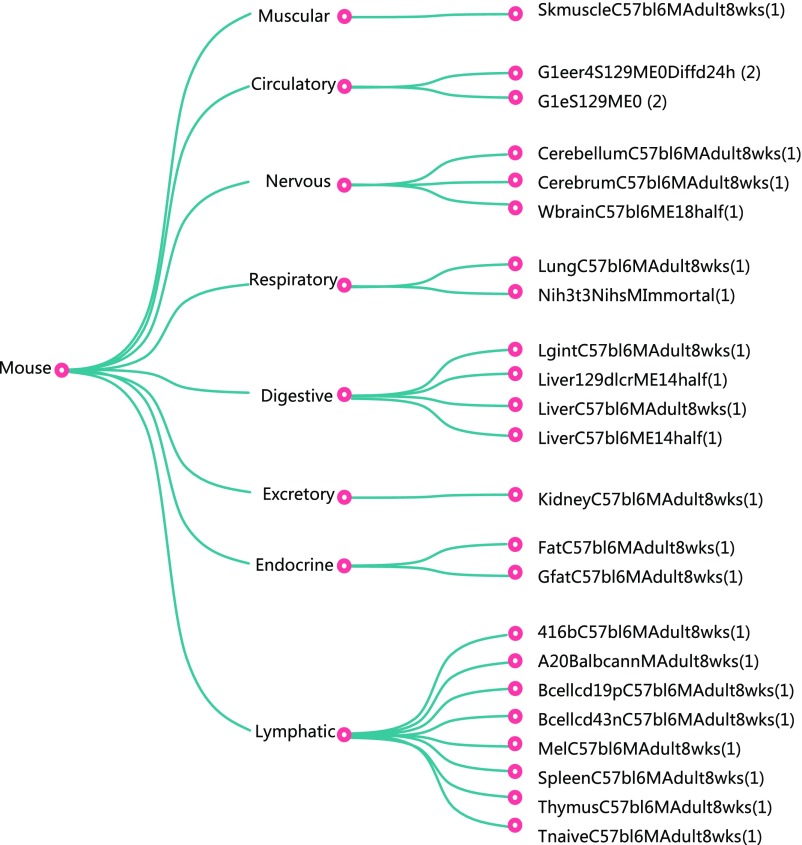
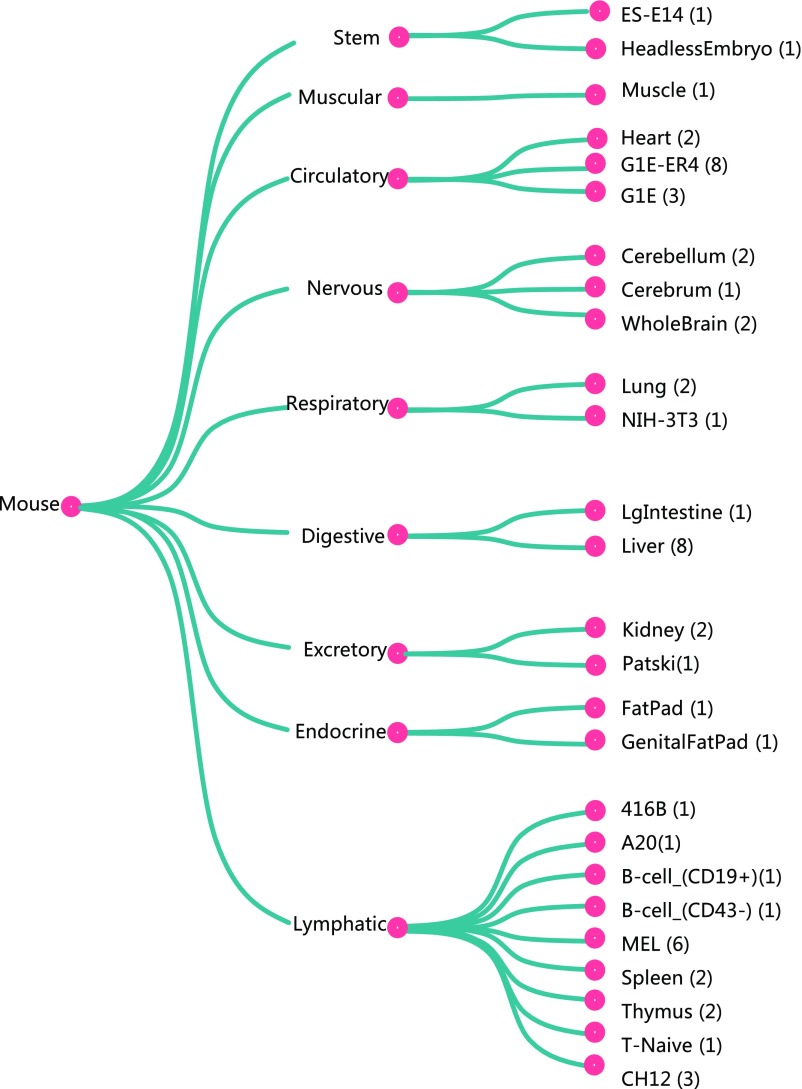
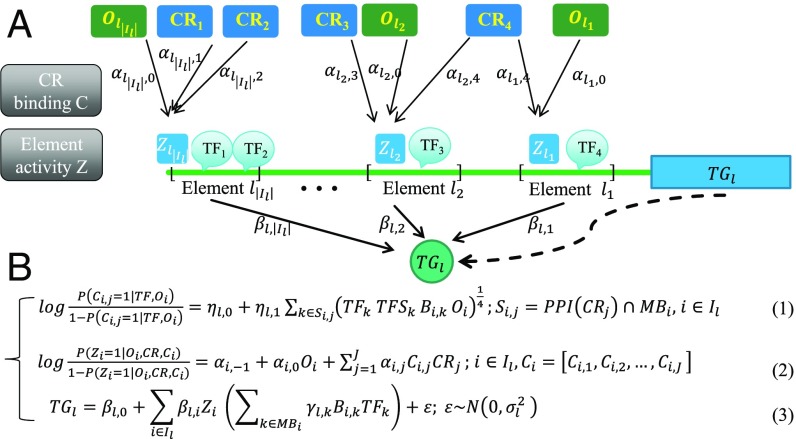
The rapid increase of genome-wide datasets on gene expression, chromatin states, and transcription factor (TF) binding locations offers an exciting opportunity to interpret the information encoded in genomes and epigenomes. This task can be challenging as it requires joint modeling of context-specific activation of cis-regulatory elements (REs) and the effects on transcription of associated regulatory factors. To meet this challenge, we propose a statistical approach based on paired expression and chromatin accessibility (PECA) data across diverse cellular contexts. In our approach, we model (i) the localization to REs of chromatin regulators (CRs) based on their interaction with sequence-specific TFs, (ii) the activation of REs due to CRs that are localized to them, and (iii) the effect of TFs bound to activated REs on the transcription of target genes (TGs). The transcriptional regulatory network inferred by PECA provides a detailed view of how trans- and cis-regulatory elements work together to affect gene expression in a context-specific manner. We illustrate the feasibility of this approach by analyzing paired expression and accessibility data from the mouse Encyclopedia of DNA Elements (ENCODE) and explore various applications of the resulting model.
Keywords: chromatin activity; chromatin regulator; gene regulation; regulatory element; transcription factor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
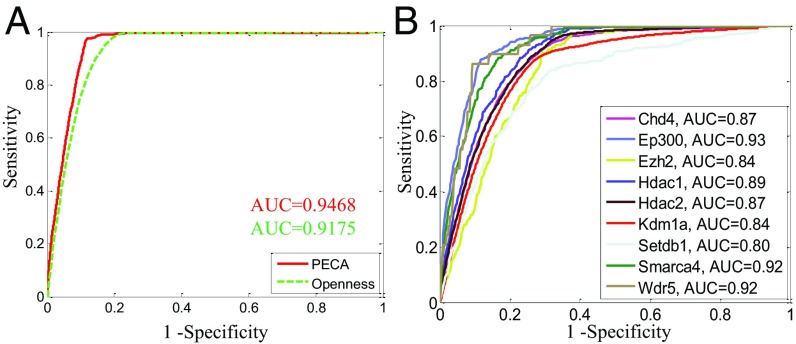
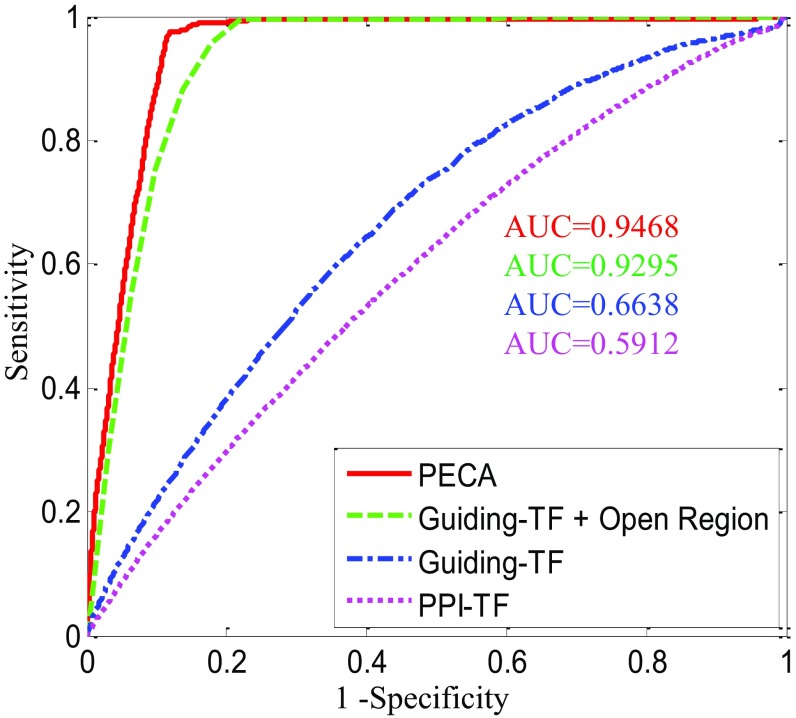
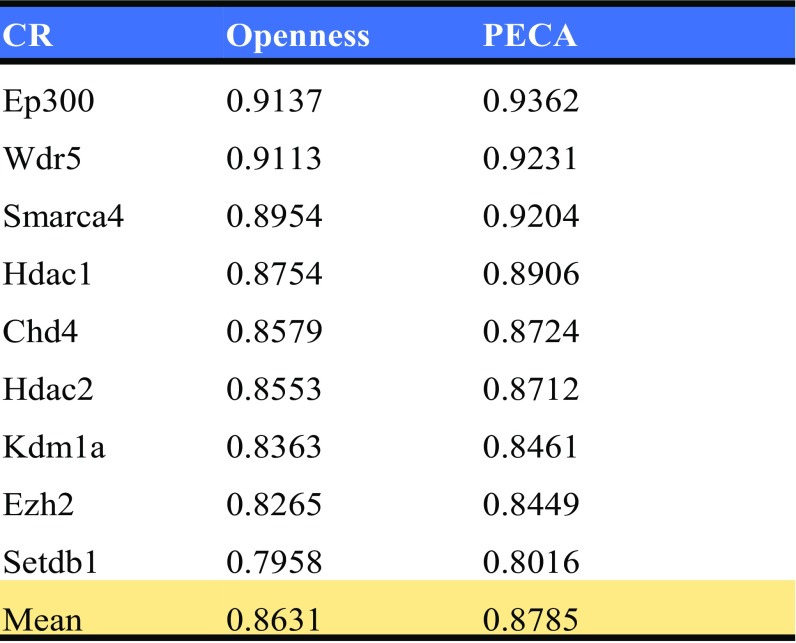
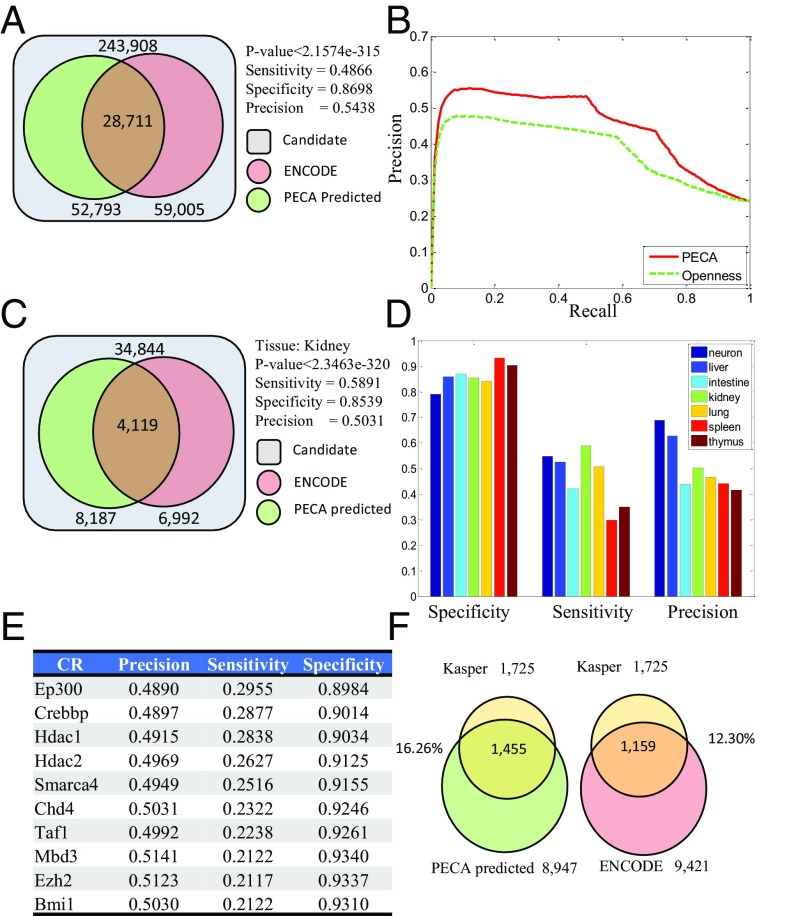
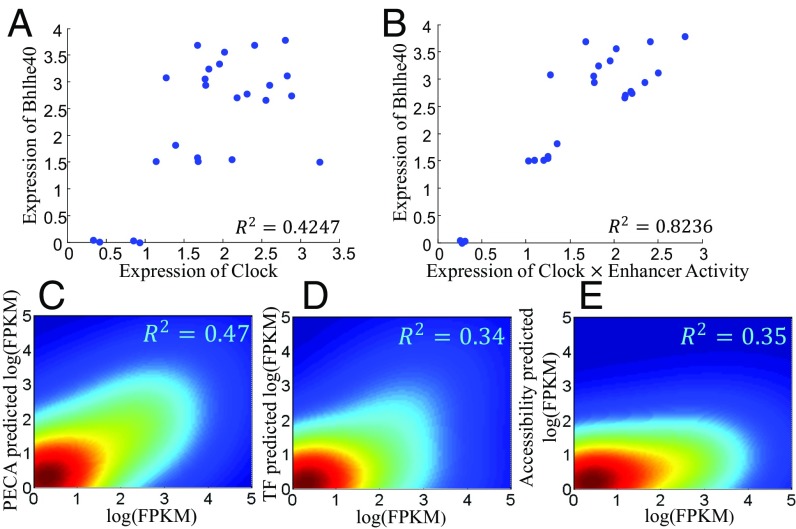
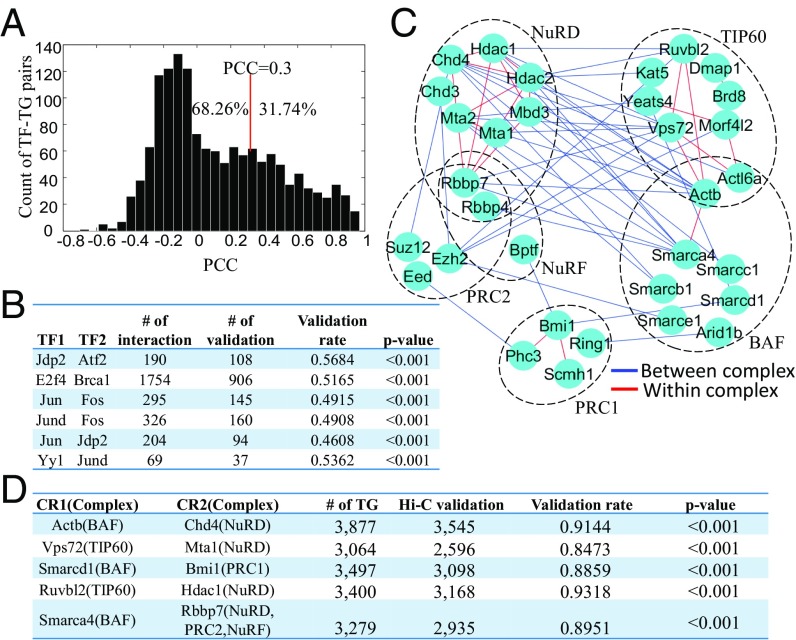
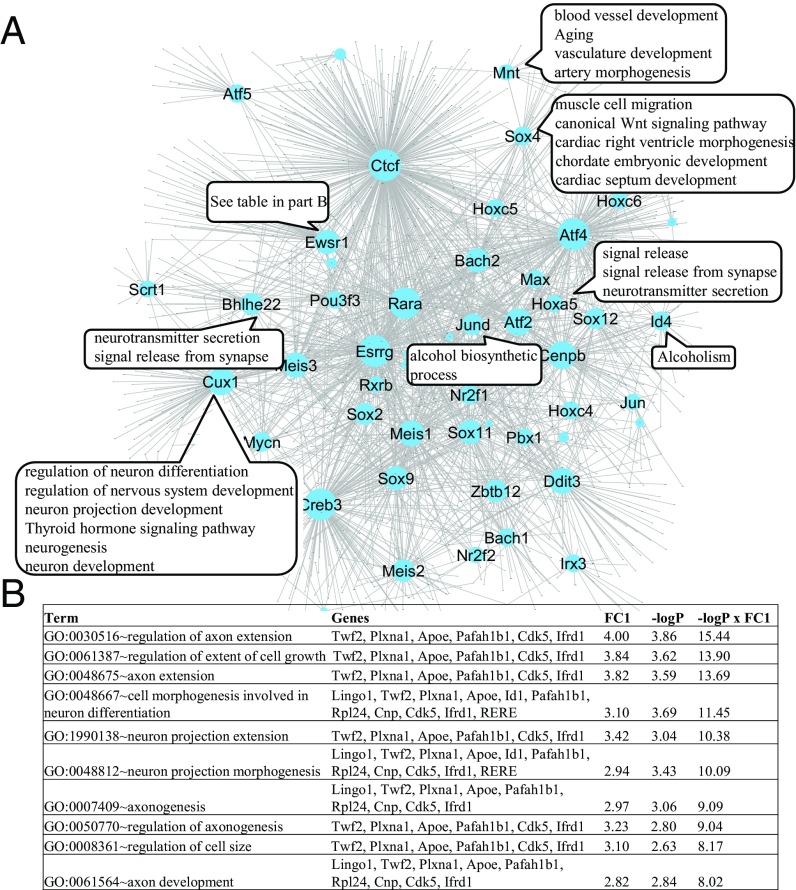
References
-
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. - PubMed
-
- Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. - PubMed
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
