

Mitochondria-meditated pathways of organ failure upon inflammation
- PMID: 28578275
- PMCID: PMC5458092
- DOI: 10.1016/j.redox.2017.05.017
Mitochondria-meditated pathways of organ failure upon inflammation
Abstract
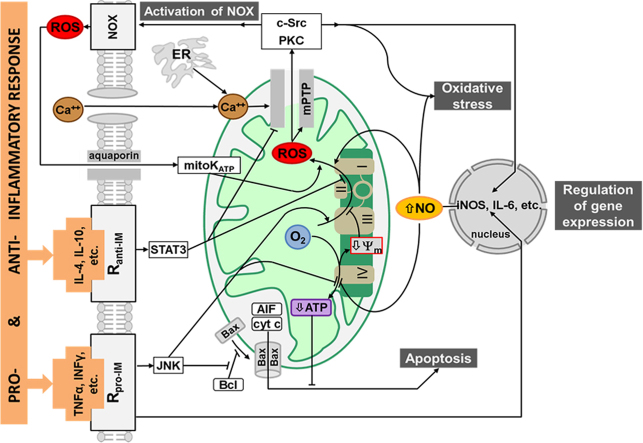
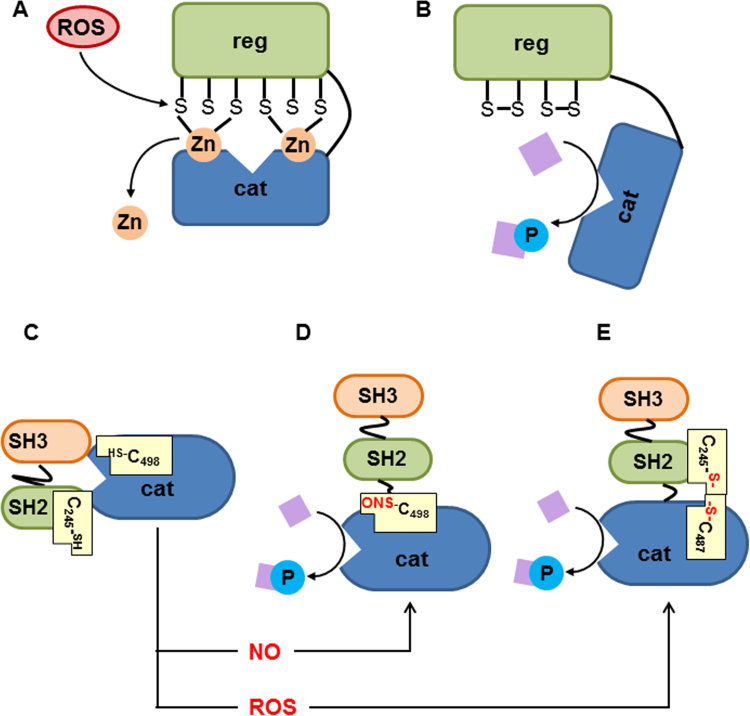
Liver failure induced by systemic inflammatory response (SIRS) is often associated with mitochondrial dysfunction but the mechanism linking SIRS and mitochondria-mediated liver failure is still a matter of discussion. Current hypotheses suggest that causative events could be a drop in ATP synthesis, opening of mitochondrial permeability transition pore, specific changes in mitochondrial morphology, impaired Ca2+ uptake, generation of mitochondrial reactive oxygen species (mtROS), turnover of mitochondria and imbalance in electron supply to the respiratory chain. The aim of this review is to critically analyze existing hypotheses, in order to highlight the most promising research lines helping to prevent liver failure induced by SIRS. Evaluation of the literature shows that there is no consistent support that impaired Ca++ metabolism, electron transport chain function and ultrastructure of mitochondria substantially contribute to liver failure. Moreover, our analysis suggests that the drop in ATP levels has protective rather than a deleterious character. Recent data suggest that the most critical mitochondrial event occurring upon SIRS is the release of mtROS in cytoplasm, which can activate two specific intracellular signaling cascades. The first is the mtROS-mediated activation of NADPH-oxidase in liver macrophages and endothelial cells; the second is the acceleration of the expression of inflammatory genes in hepatocytes. The signaling action of mtROS is strictly controlled in mitochondria at three points, (i) at the site of ROS generation at complex I, (ii) the site of mtROS release in cytoplasm via permeability transition pore, and (iii) interaction with specific kinases in cytoplasm. The systems controlling mtROS-signaling include pro- and anti-inflammatory mediators, nitric oxide, Ca2+ and NADPH-oxidase. Analysis of the literature suggests that further research should be focused on the impact of mtROS on organ failure induced by inflammation and simultaneously providing a new theoretical basis for a targeted therapy of overwhelmed inflammatory response.
Keywords: Inflammation; Liver failure; Mitochondria; Reactive oxygen species; Signaling.
Copyright © 2017 The Authors. Published by Elsevier B.V. All rights reserved.
Figures



Similar articles
-
Vicious inducible nitric oxide synthase-mitochondrial reactive oxygen species cycle accelerates inflammatory response and causes liver injury in rats.Antioxid Redox Signal. 2015 Mar 1;22(7):572-86. doi: 10.1089/ars.2014.5996. Epub 2014 Dec 22. Antioxid Redox Signal. 2015. PMID: 25365698
-
Proton leak regulates mitochondrial reactive oxygen species generation in endothelial cell activation and inflammation - A novel concept.Arch Biochem Biophys. 2019 Feb 15;662:68-74. doi: 10.1016/j.abb.2018.12.002. Epub 2018 Dec 3. Arch Biochem Biophys. 2019. PMID: 30521782 Free PMC article. Review.
-
Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells.Can J Physiol Pharmacol. 2017 Mar;95(3):247-252. doi: 10.1139/cjpp-2016-0515. Epub 2016 Nov 5. Can J Physiol Pharmacol. 2017. PMID: 27925481 Free PMC article. Review.
-
Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils.Biochim Biophys Acta Mol Basis Dis. 2020 May 1;1866(5):165664. doi: 10.1016/j.bbadis.2020.165664. Epub 2020 Jan 8. Biochim Biophys Acta Mol Basis Dis. 2020. PMID: 31926265
-
Molecular hydrogen inhibits lipopolysaccharide-triggered NLRP3 inflammasome activation in macrophages by targeting the mitochondrial reactive oxygen species.Biochim Biophys Acta. 2016 Jan;1863(1):50-5. doi: 10.1016/j.bbamcr.2015.10.012. Epub 2015 Oct 18. Biochim Biophys Acta. 2016. PMID: 26488087
Cited by
-
Zaluzanin C Alleviates Inflammation and Lipid Accumulation in Kupffer Cells and Hepatocytes by Regulating Mitochondrial ROS.Molecules. 2023 Nov 8;28(22):7484. doi: 10.3390/molecules28227484. Molecules. 2023. PMID: 38005205 Free PMC article.
-
Emerging perspectives on mitochondrial dysfunction and inflammation in Alzheimer's disease.BMB Rep. 2020 Jan;53(1):35-46. doi: 10.5483/BMBRep.2020.53.1.274. BMB Rep. 2020. PMID: 31818363 Free PMC article. Review.
-
Mitochondria-Targeted Antioxidants SkQ1 and MitoTEMPO Failed to Exert a Long-Term Beneficial Effect in Murine Polymicrobial Sepsis.Oxid Med Cell Longev. 2017;2017:6412682. doi: 10.1155/2017/6412682. Epub 2017 Sep 19. Oxid Med Cell Longev. 2017. PMID: 29104729 Free PMC article.
-
A Review of Antibiotic Efficacy in COVID-19 Control.J Immunol Res. 2023 Oct 10;2023:6687437. doi: 10.1155/2023/6687437. eCollection 2023. J Immunol Res. 2023. PMID: 37854054 Free PMC article. Review.
-
Suxiao Jiuxin Pill protects cardiomyocytes against mitochondrial injury and alters gene expression during ischemic injury.Exp Ther Med. 2017 Oct;14(4):3523-3532. doi: 10.3892/etm.2017.4964. Epub 2017 Aug 18. Exp Ther Med. 2017. PMID: 29042943 Free PMC article.
References
-
- Bone R.C. Toward an epidemiology and natural history of SIRS (systemic inflammatory response syndrome) J. Am. Med. Assoc. 1992;268:3452–3455. - PubMed
-
- Levy M., Fink M., Marshall J., Abraham E., Angus D., Cook D. SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Crit. Care Med. 2001;31(2003):1250–1256. - PubMed
-
- Brealey D., Brand M., Hargreaves I., Heales S., Land J., Smolenski R. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
