

How the Same Core Catalytic Machinery Catalyzes 17 Different Reactions: the Serine-Histidine-Aspartate Catalytic Triad of α/β-Hydrolase Fold Enzymes
- PMID: 28580193
- PMCID: PMC5455348
- DOI: 10.1021/acscatal.5b01539
How the Same Core Catalytic Machinery Catalyzes 17 Different Reactions: the Serine-Histidine-Aspartate Catalytic Triad of α/β-Hydrolase Fold Enzymes
Abstract
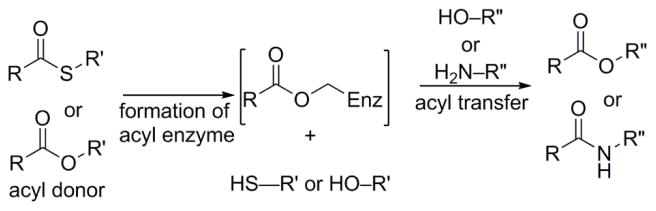
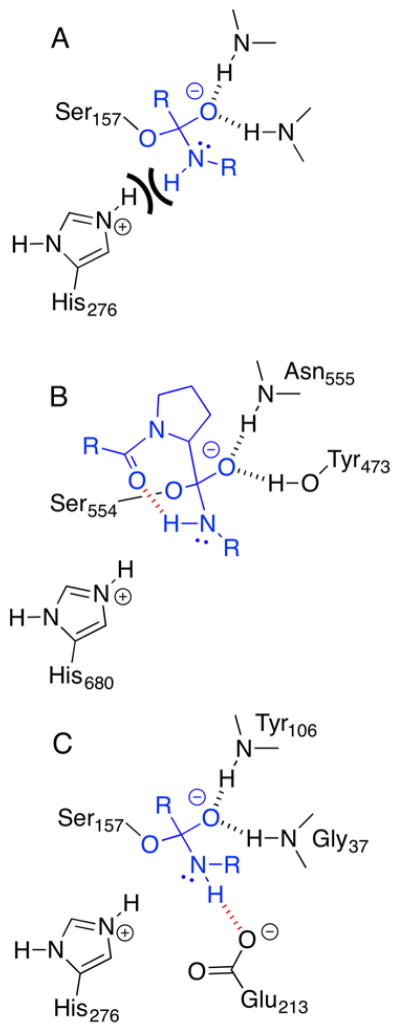
Enzymes within a family often catalyze different reactions. In some cases, this variety stems from different catalytic machinery, but in other cases the machinery is identical; nevertheless, the enzymes catalyze different reactions. In this review, we examine the subset of α/β-hydrolase fold enzymes that contain the serine-histidine-aspartate catalytic triad. In spite of having the same protein fold and the same core catalytic machinery, these enzymes catalyze seventeen different reaction mechanisms. The most common reactions are hydrolysis of C-O, C-N and C-C bonds (Enzyme Classification (EC) group 3), but other enzymes are oxidoreductases (EC group 1), acyl transferases (EC group 2), lyases (EC group 4) or isomerases (EC group 5). Hydrolysis reactions often follow the canonical esterase mechanism, but eight variations occur where either the formation or cleavage of the acyl enzyme intermediate differs. The remaining eight mechanisms are lyase-type elimination reactions, which do not have an acyl enzyme intermediate and, in four cases, do not even require the catalytic serine. This diversity of mechanisms from the same catalytic triad stems from the ability of the enzymes to bind different substrates, from the requirements for different chemical steps imposed by these new substrates and, only in about half of the cases, from additional hydrogen bond partners or additional general acids/bases in the active site. This detailed analysis shows that binding differences and non-catalytic residues create new mechanisms and are essential for understanding and designing efficient enzymes.
Keywords: X-ray structures; catalytic triad; divergent evolution; hydrolase; lyase; mechanism; oxyanion hole.
Figures


































References
-
- Albery W, Knowles J. Biochemistry. 1976;15:5631–5640. - PubMed
-
- Wolfenden R. Chem Rev. 2006;106:3379–3396. - PubMed
-
- Kiss G, Çelebi-Ölçüm N, Moretti R, Baker D, Houk KN. Angew Chem Int Ed. 2013;52:5700–5725. - PubMed
-
-
For example, Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ. Science. 2010;329:305–309.
-
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases