

The evolution and population diversity of human-specific segmental duplications
- PMID: 28580430
- PMCID: PMC5450946
- DOI: 10.1038/s41559-016-0069
The evolution and population diversity of human-specific segmental duplications
Abstract
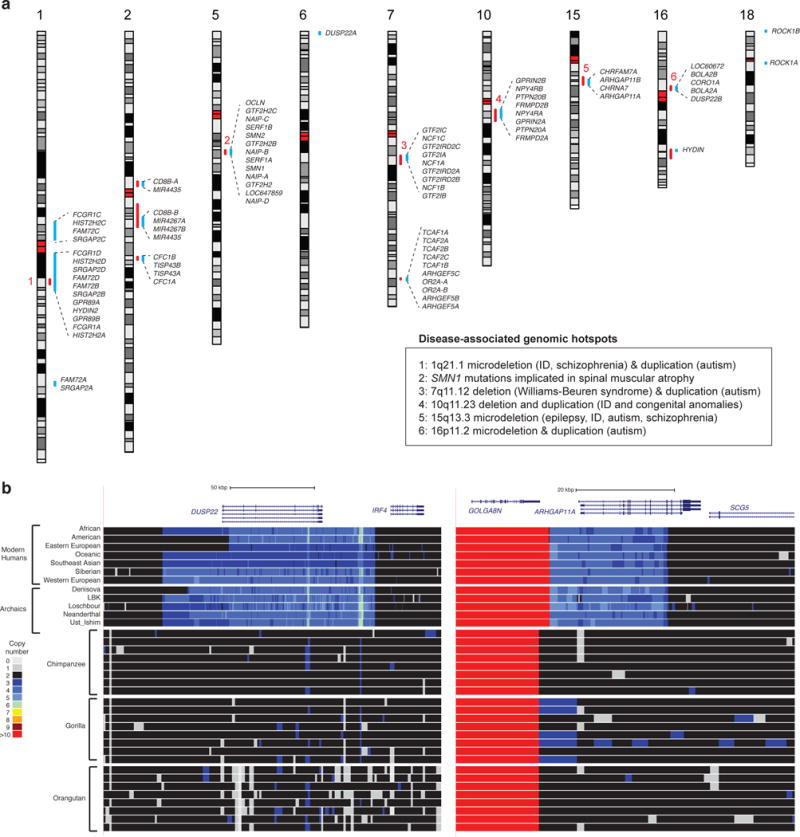
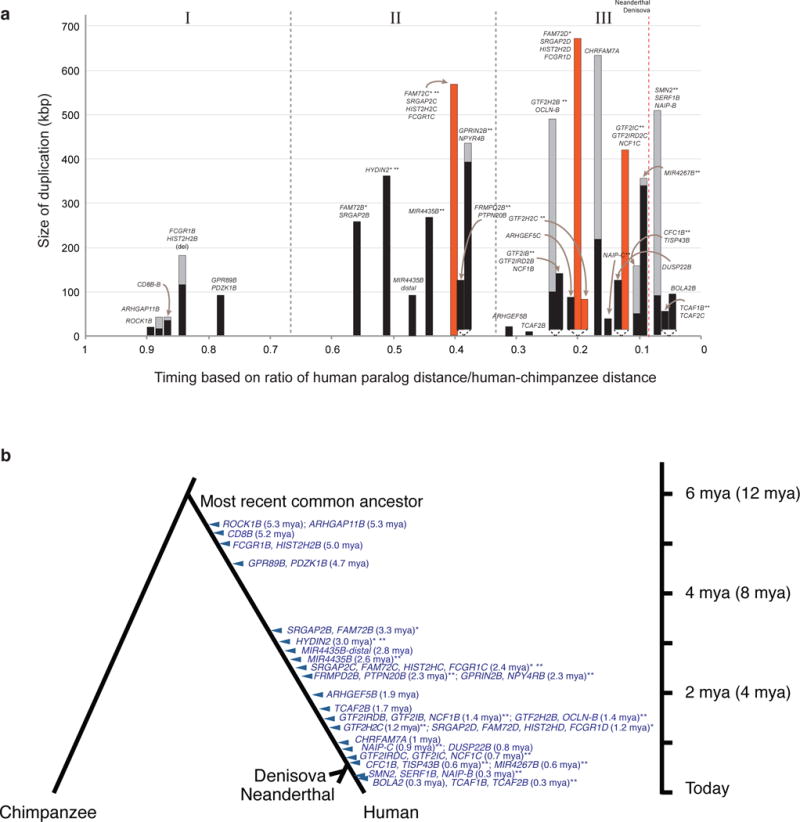
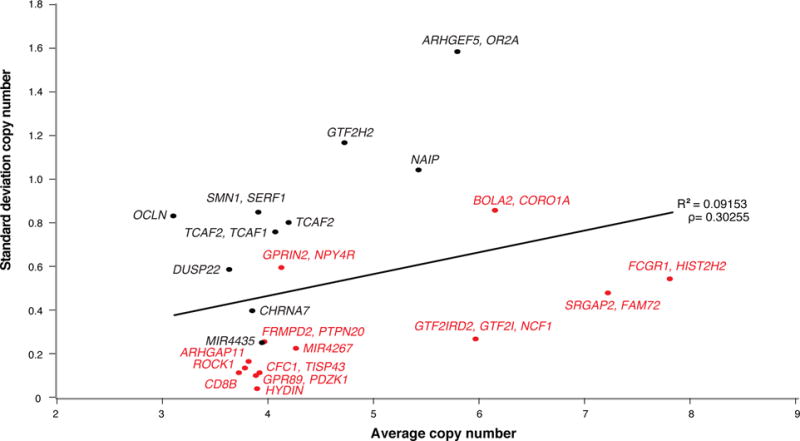
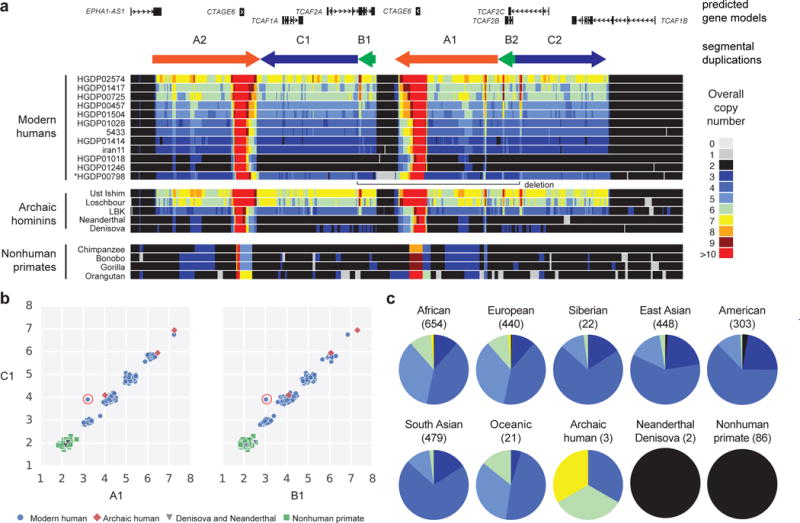
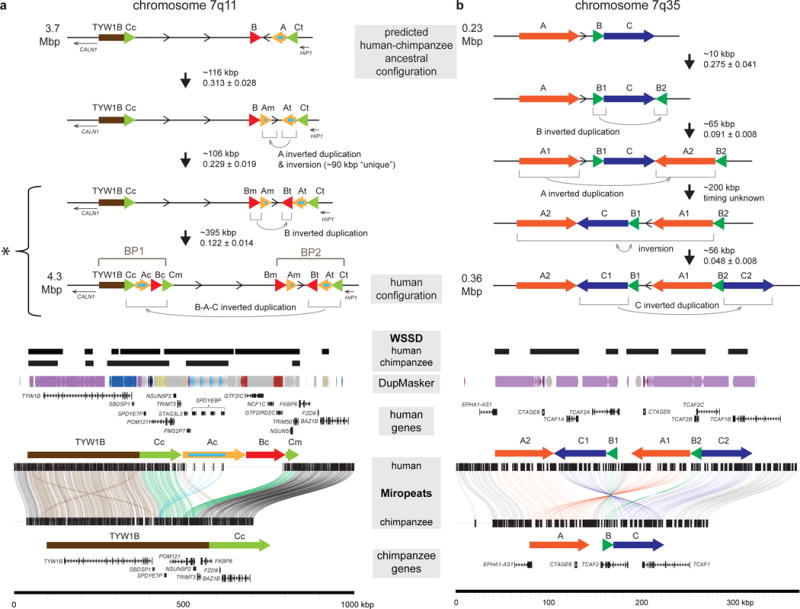
Segmental duplications contribute to human evolution, adaptation and genomic instability but are often poorly characterized. We investigate the evolution, genetic variation and coding potential of human-specific segmental duplications (HSDs). We identify 218 HSDs based on analysis of 322 deeply sequenced archaic and contemporary hominid genomes. We sequence 550 human and nonhuman primate genomic clones to reconstruct the evolution of the largest, most complex regions with protein-coding potential (n=80 genes/33 gene families). We show that HSDs are non-randomly organized, associate preferentially with ancestral ape duplications termed "core duplicons", and evolved primarily in an interspersed inverted orientation. In addition to Homo sapiens-specific gene expansions (e.g., TCAF1/2), we highlight ten gene families (e.g., ARHGAP11B and SRGAP2C) where copy number never returns to the ancestral state, there is evidence of mRNA splicing, and no common gene-disruptive mutations are observed in the general population. Such duplicates are candidates for the evolution of human-specific adaptive traits.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc., is a consultant for Kunming University of Science and Technology (KUST) as part of the 1000 China Talent Program, and was an SAB member of Pacific Biosciences, Inc. (2009–2013).
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
