

Applications of Nonenzymatic Catalysts to the Alteration of Natural Products
- PMID: 28580785
- PMCID: PMC5742423
- DOI: 10.1021/acs.chemrev.7b00022
Applications of Nonenzymatic Catalysts to the Alteration of Natural Products
Abstract
The application of small molecules as catalysts for the diversification of natural product scaffolds is reviewed. Specifically, principles that relate to the selectivity challenges intrinsic to complex molecular scaffolds are summarized. The synthesis of analogues of natural products by this approach is then described as a quintessential "late-stage functionalization" exercise wherein natural products serve as the lead scaffolds. Given the historical application of enzymatic catalysts to the site-selective alteration of complex molecules, the focus of this Review is on the recent studies of nonenzymatic catalysts. Reactions involving hydroxyl group derivatization with a variety of electrophilic reagents are discussed. C-H bond functionalizations that lead to oxidations, aminations, and halogenations are also presented. Several examples of site-selective olefin functionalizations and C-C bond formations are also included. Numerous classes of natural products have been subjected to these studies of site-selective alteration including polyketides, glycopeptides, terpenoids, macrolides, alkaloids, carbohydrates, and others. What emerges is a platform for chemical remodeling of naturally occurring scaffolds that targets virtually all known chemical functionalities and microenvironments. However, challenges for the design of very broad classes of catalysts, with even broader selectivity demands (e.g., stereoselectivity, functional group selectivity, and site-selectivity) persist. Yet, a significant spectrum of powerful, catalytic alterations of complex natural products now exists such that expansion of scope seems inevitable. Several instances of biological activity assays of remodeled natural product derivatives are also presented. These reports may foreshadow further interdisciplinary impacts for catalytic remodeling of natural products, including contributions to SAR development, mode of action studies, and eventually medicinal chemistry.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
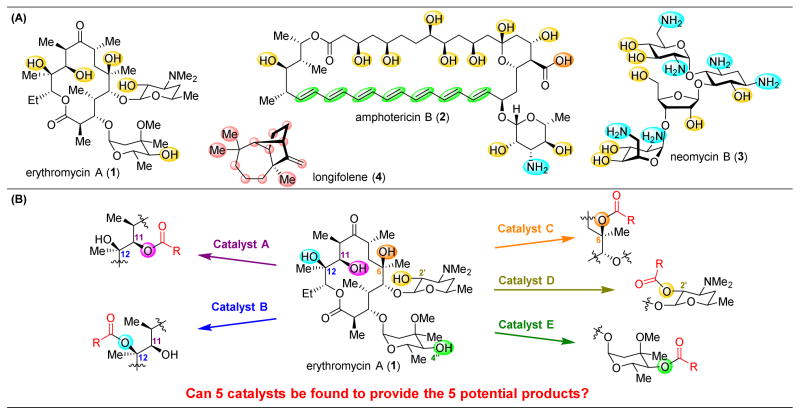
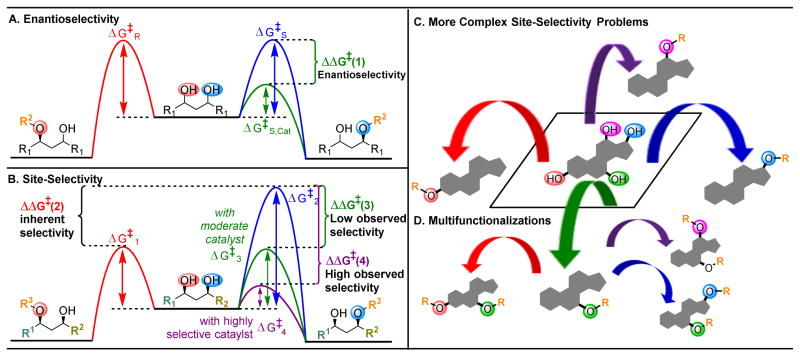
 pathway,
pathway,
 ) will result in high enantioselectivities,
(B) Energy profiles of a site-selective transformation, where reactive groups are nonequivalent. Depending on the inherent energy profiles of the functional groups (
) will result in high enantioselectivities,
(B) Energy profiles of a site-selective transformation, where reactive groups are nonequivalent. Depending on the inherent energy profiles of the functional groups (
 versus
versus
 pathways,
pathways,
 ), catalytic reduction of an energy barrier may not result in high observed selectivities (
pathway,
), catalytic reduction of an energy barrier may not result in high observed selectivities (
pathway,
 ). The achievement of highly selective functionalizations may require substantially more selective catalysts (
). The achievement of highly selective functionalizations may require substantially more selective catalysts (
 pathway,
pathway,
 ). (C) This problem is compounded by the addition of more reactive groups and (D) the ability for substrates to undergo multiple derivatization events.
). (C) This problem is compounded by the addition of more reactive groups and (D) the ability for substrates to undergo multiple derivatization events.
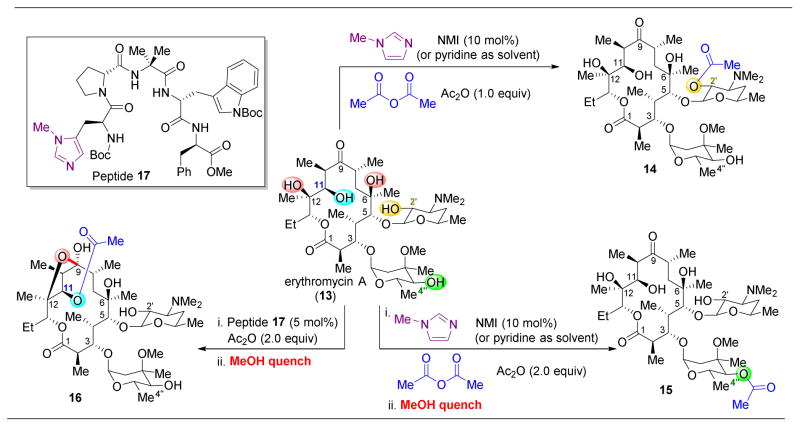
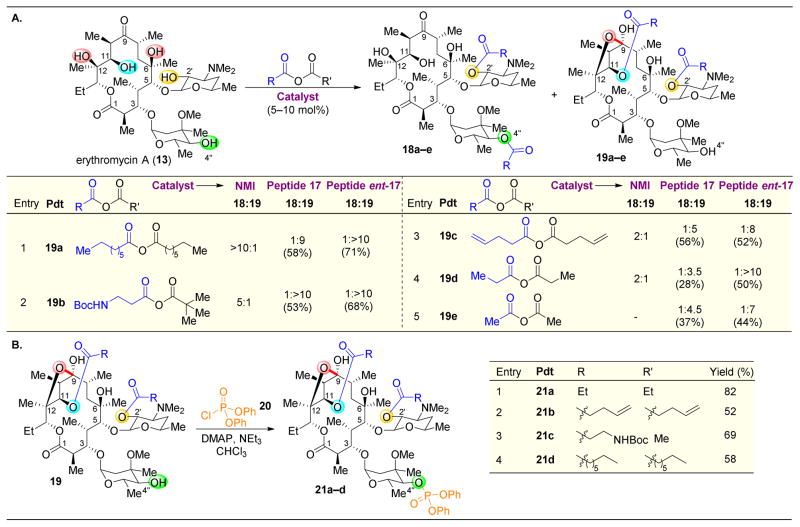
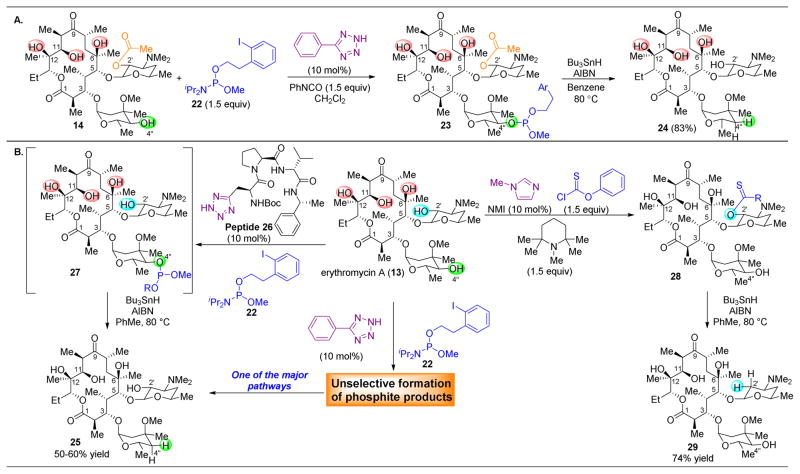
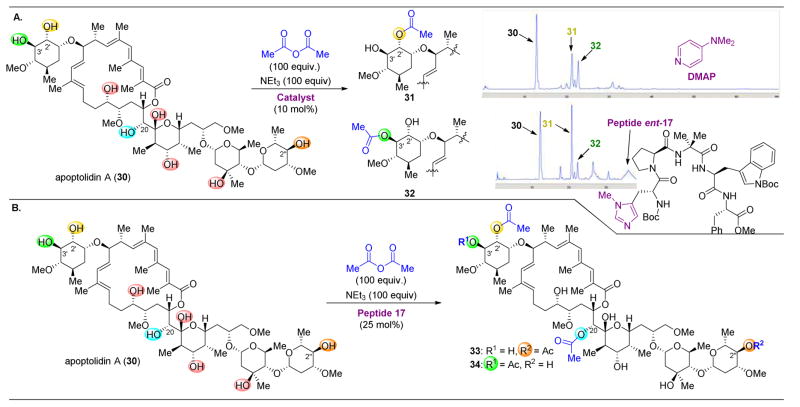
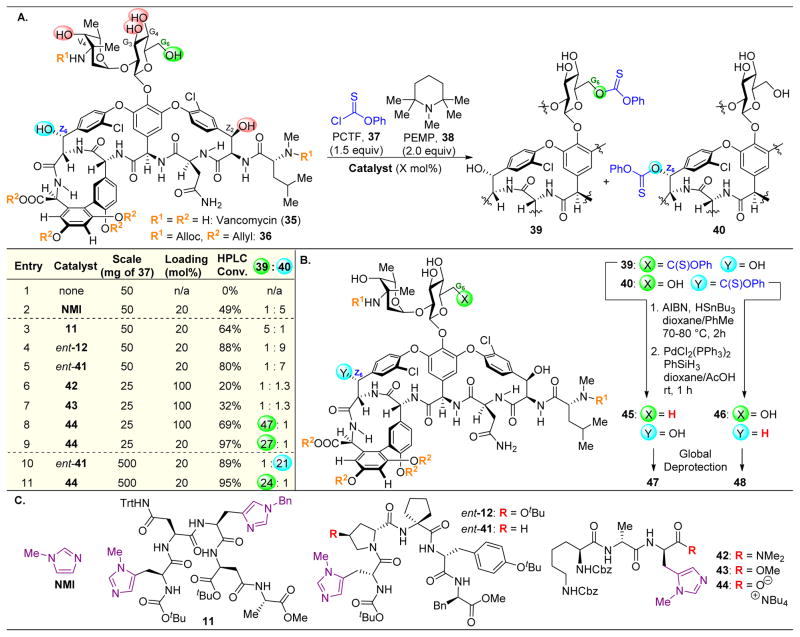
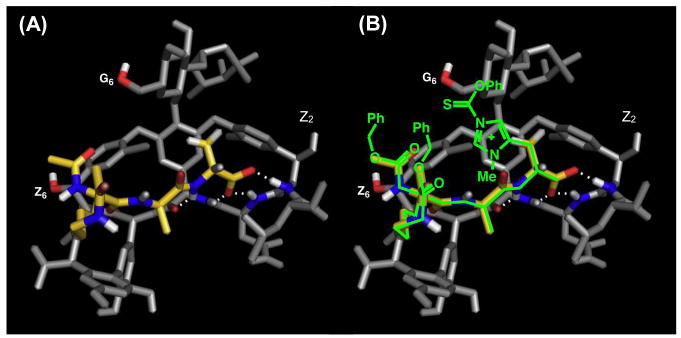
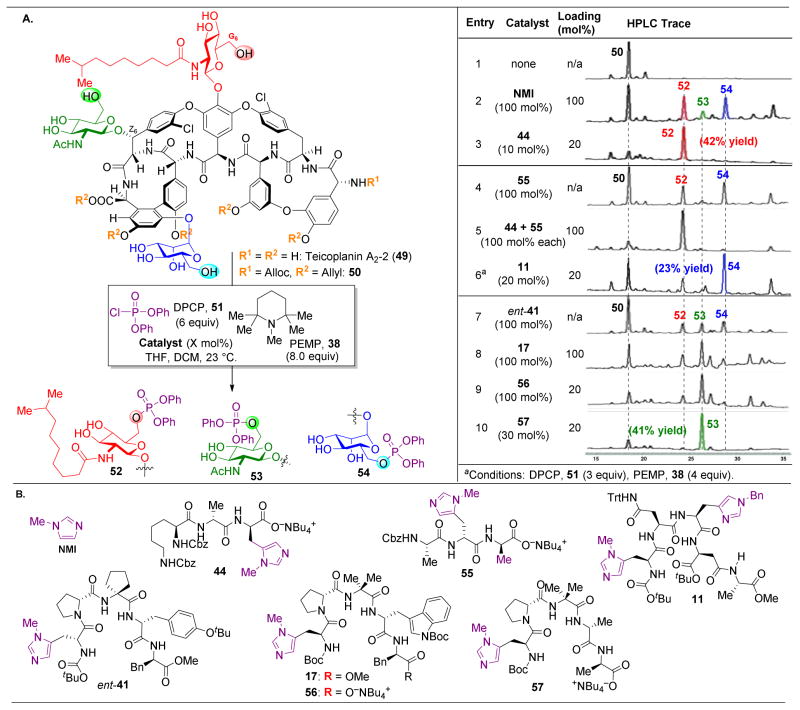
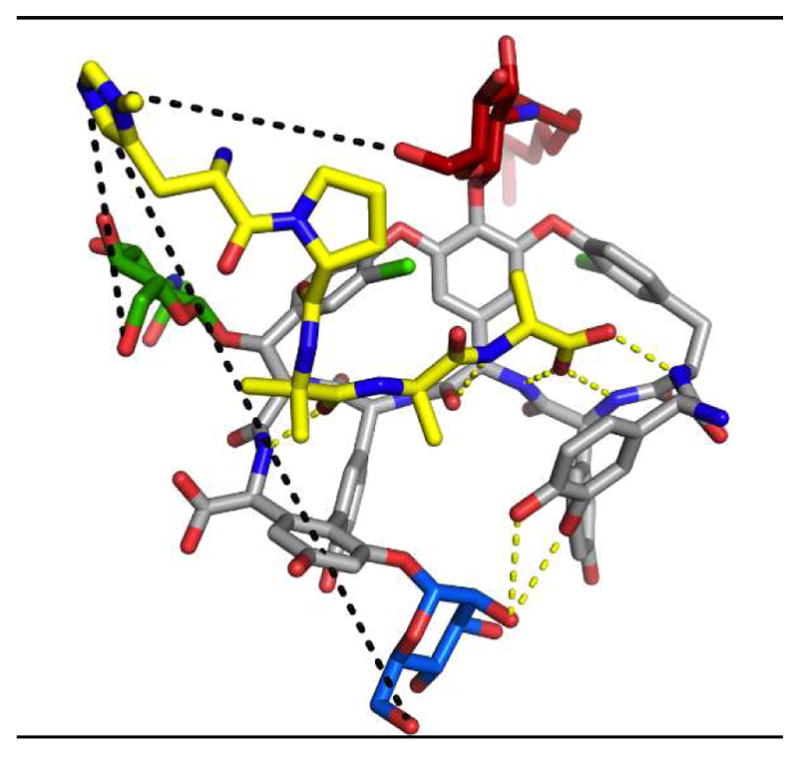
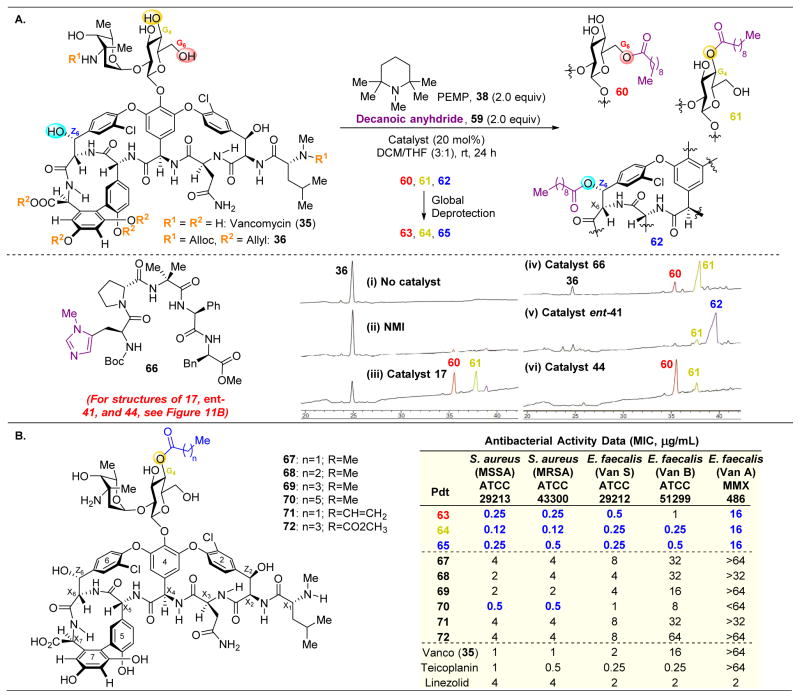
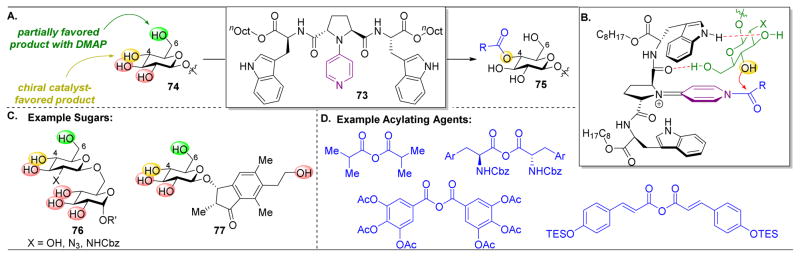
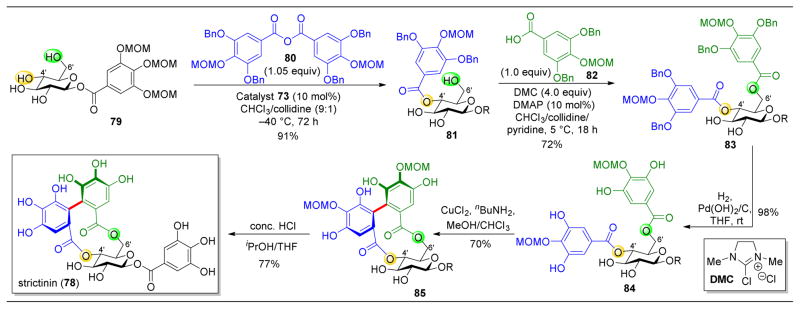
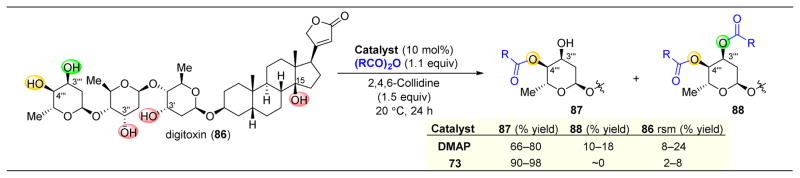
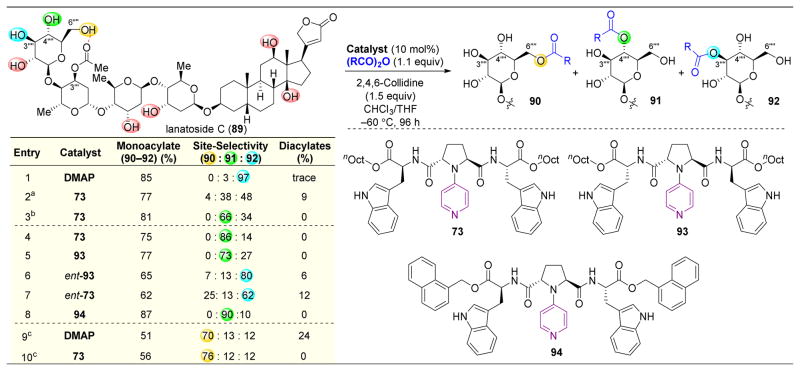
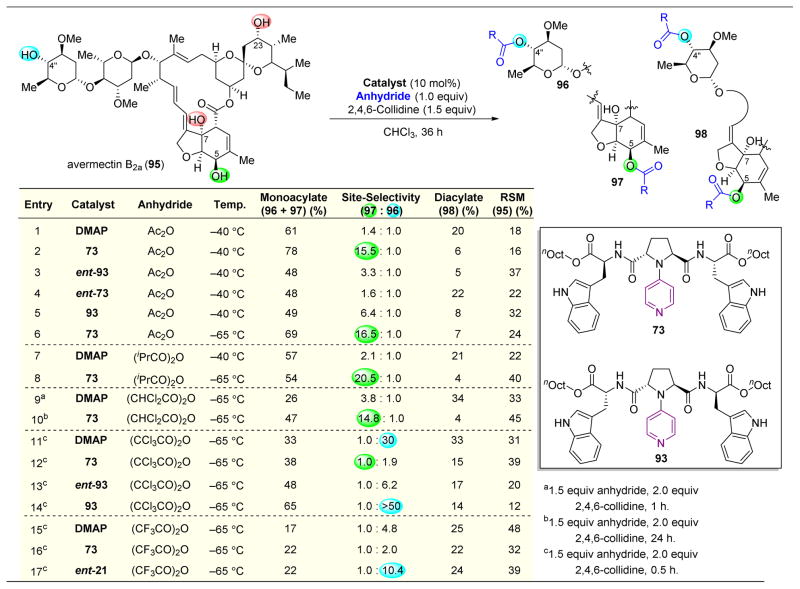
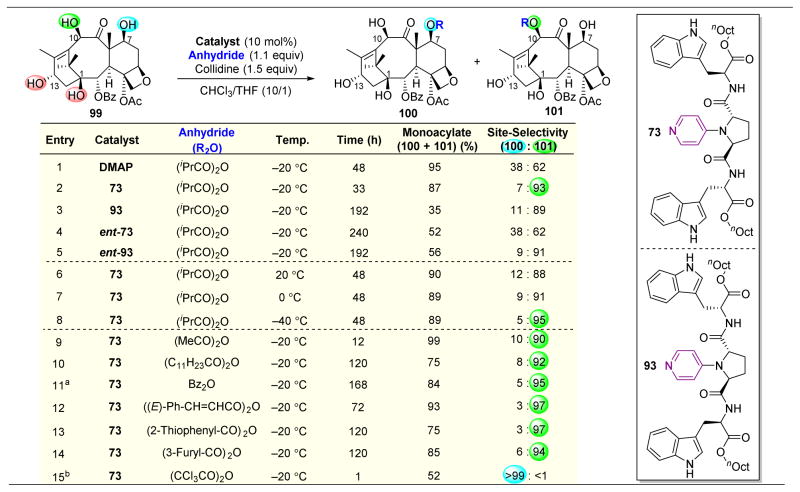
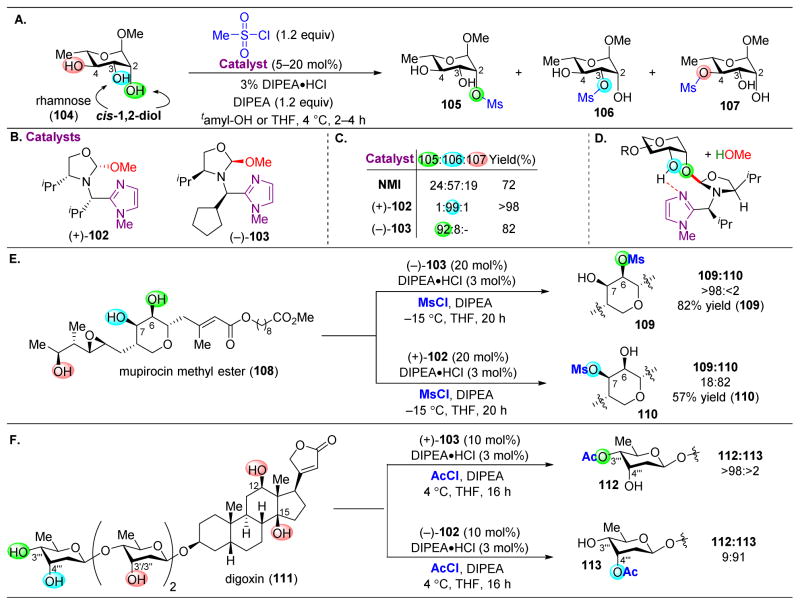
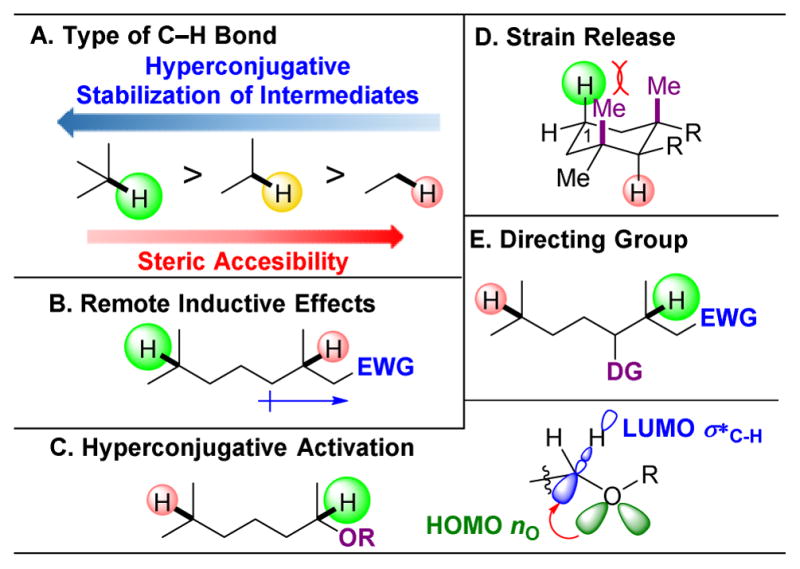
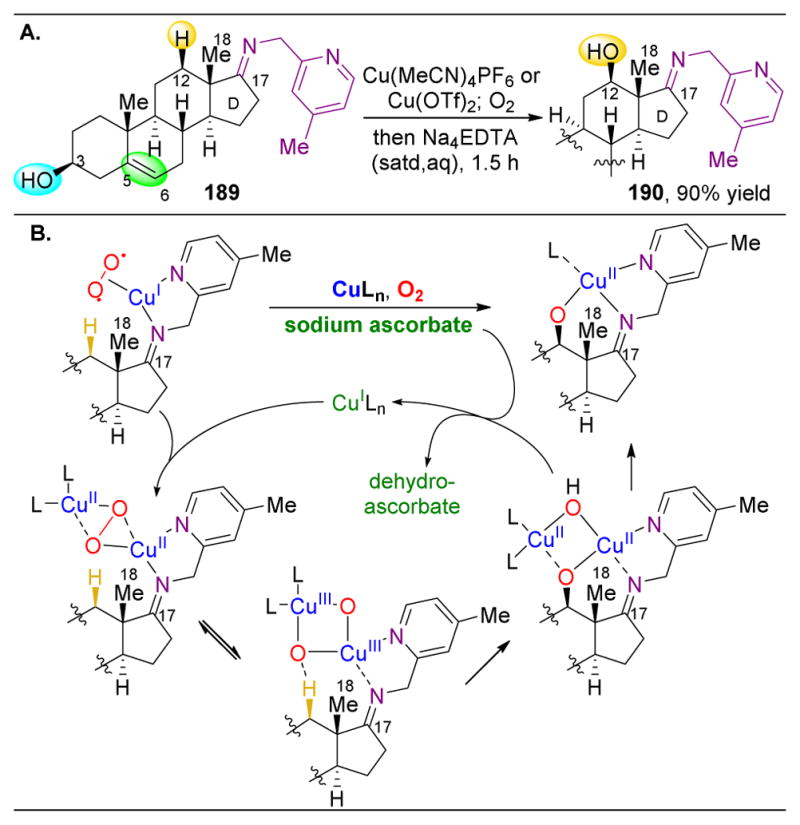
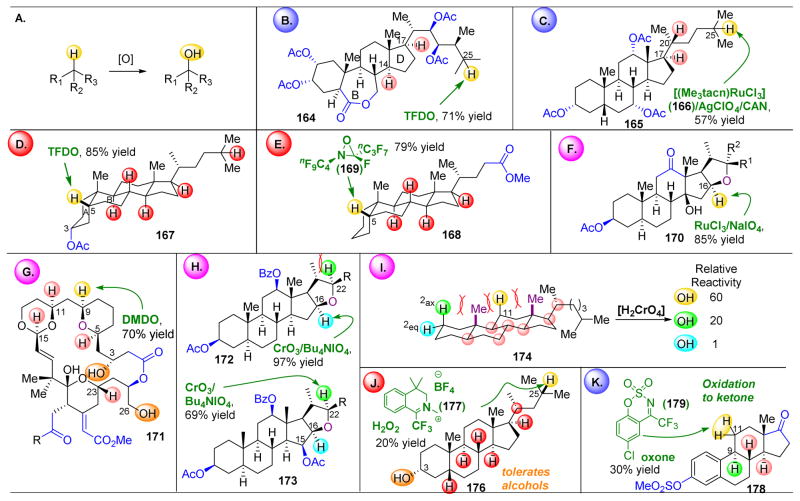 ,
, and
orbs represent dominance of electronic, steric, and stereoelectronic factors respectively.,,–
,
, and
orbs represent dominance of electronic, steric, and stereoelectronic factors respectively.,,–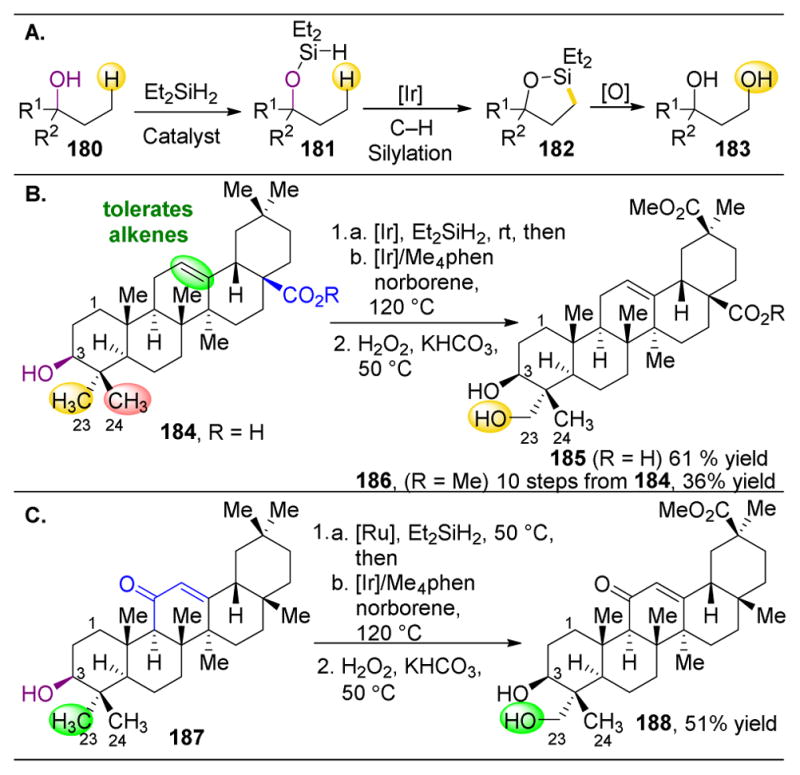
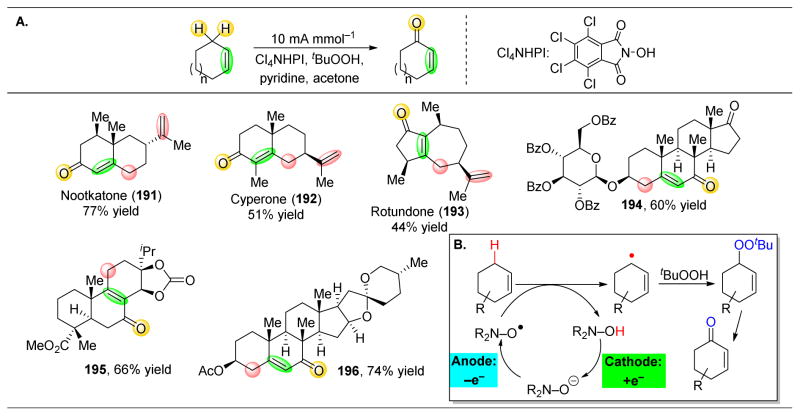
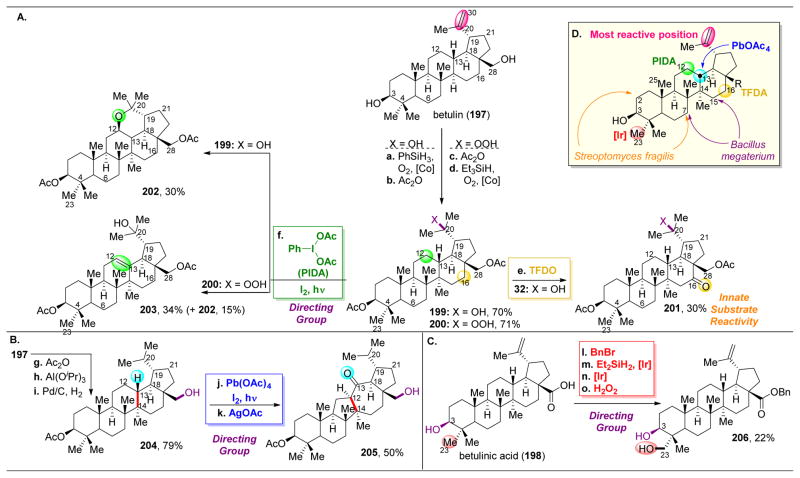
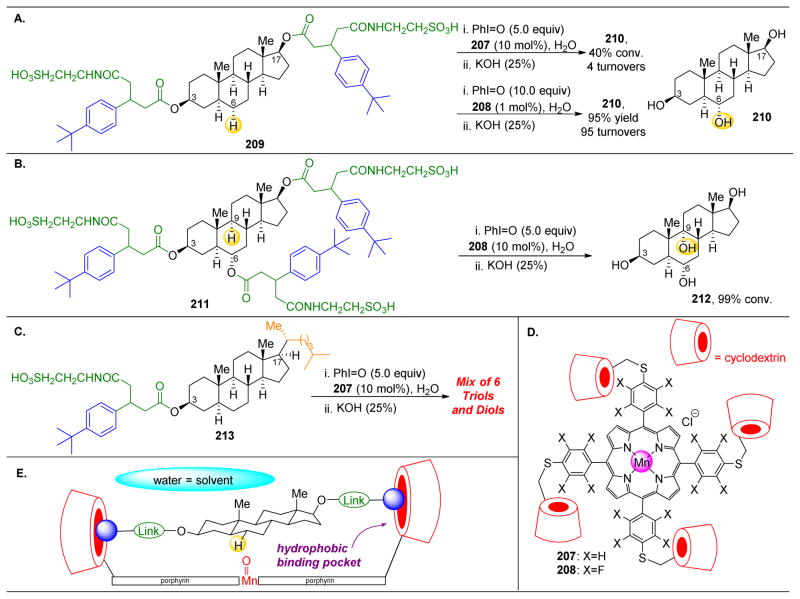
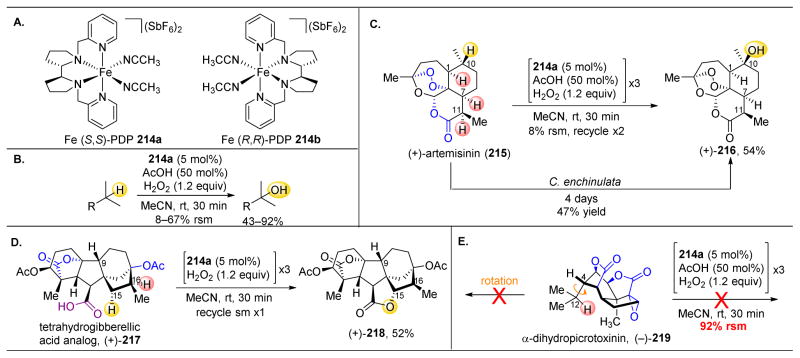
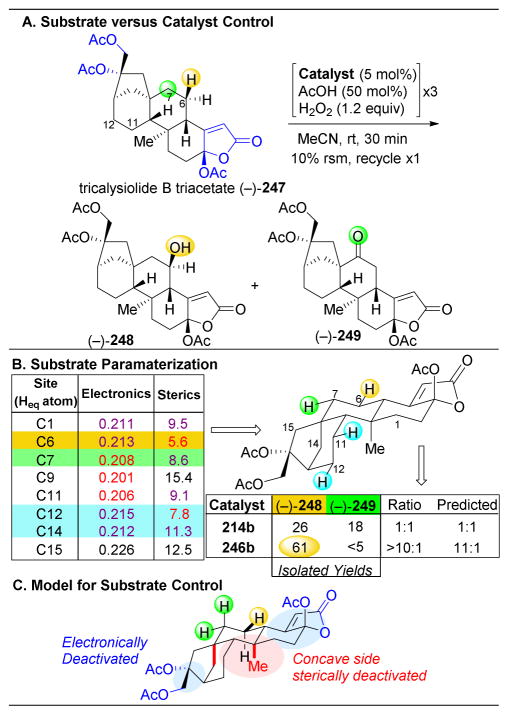
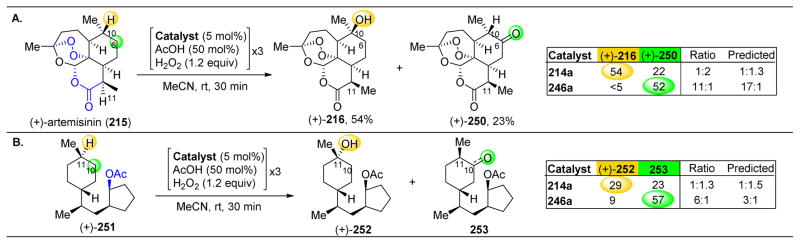
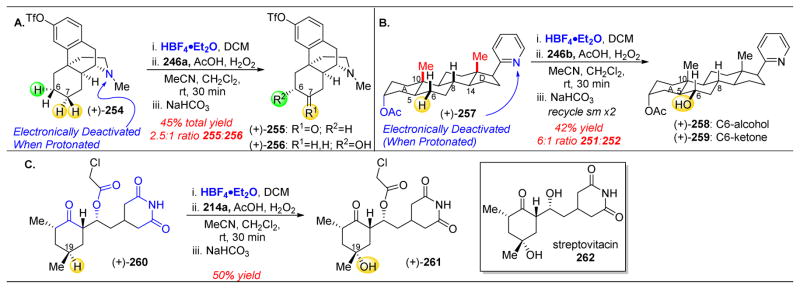
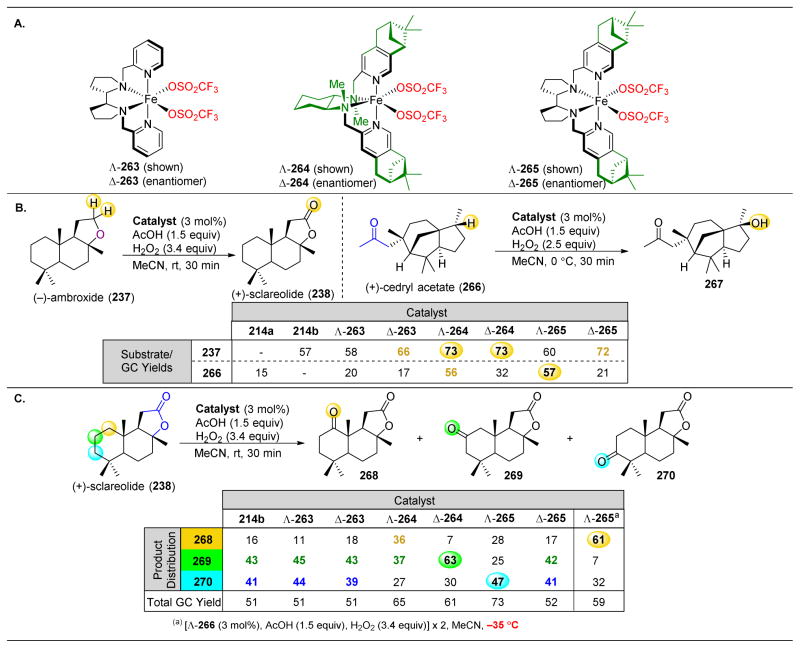
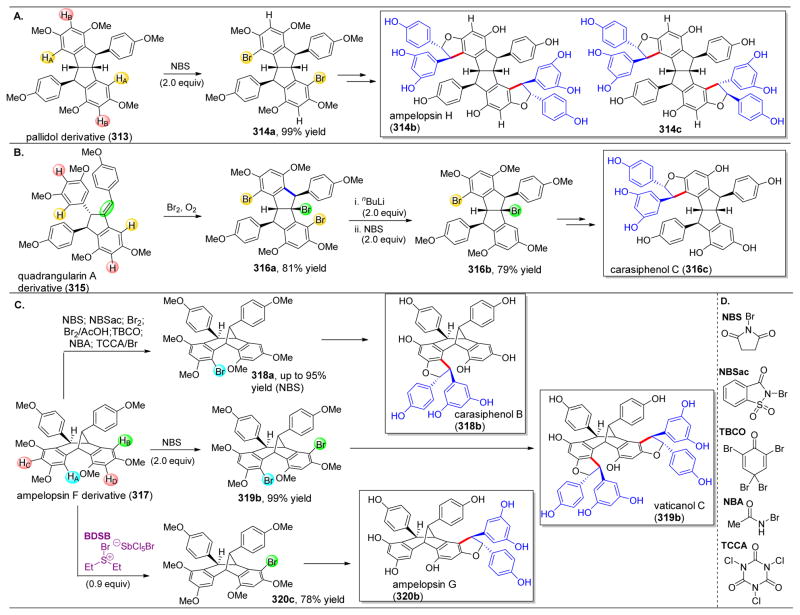
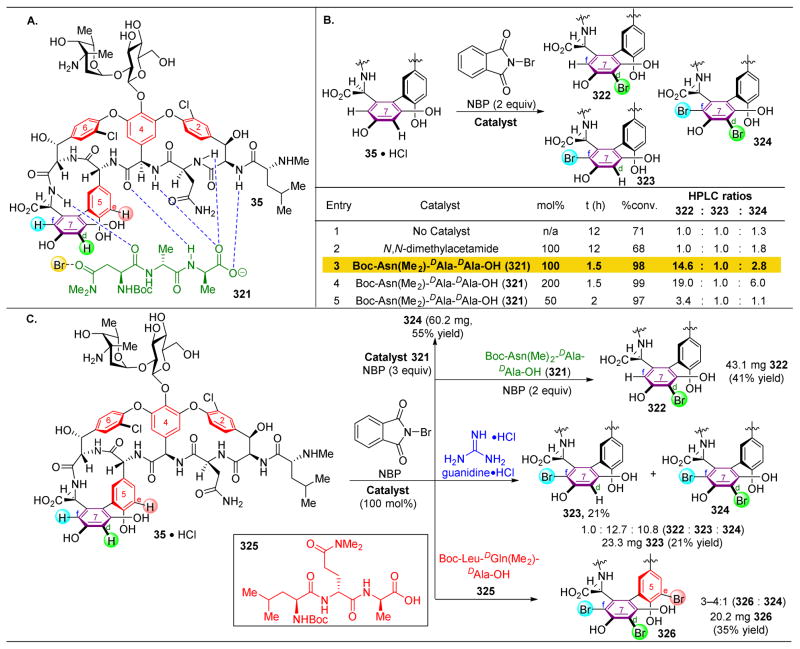
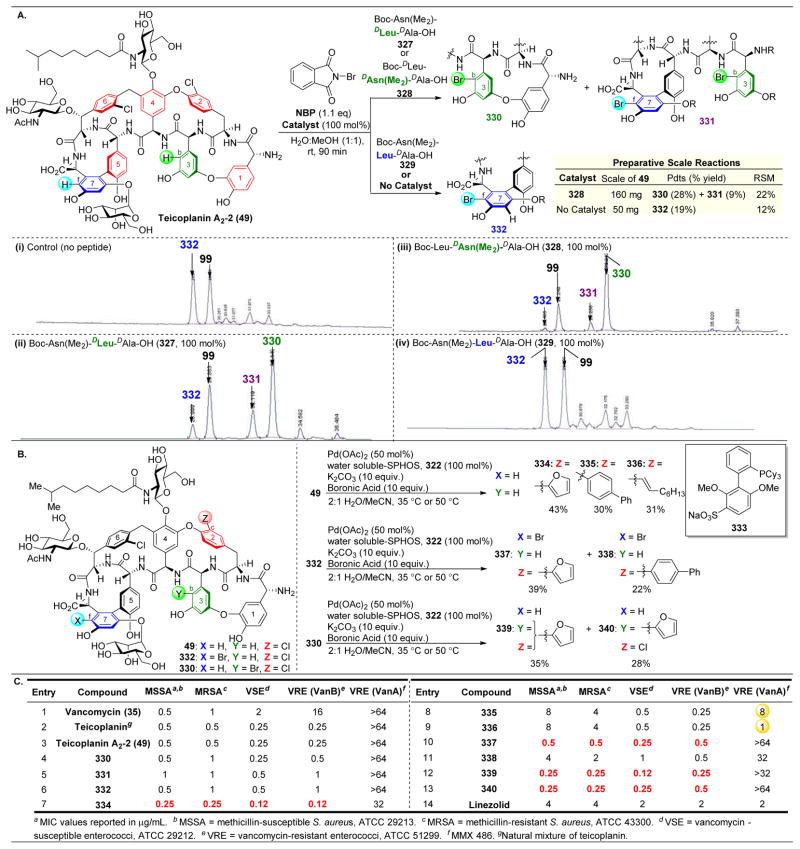
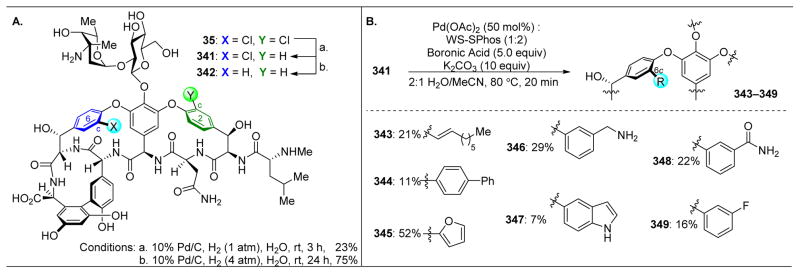
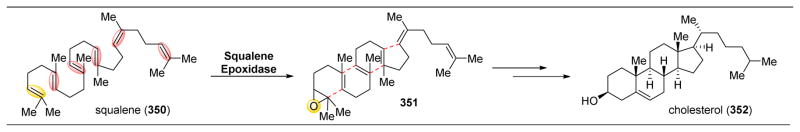
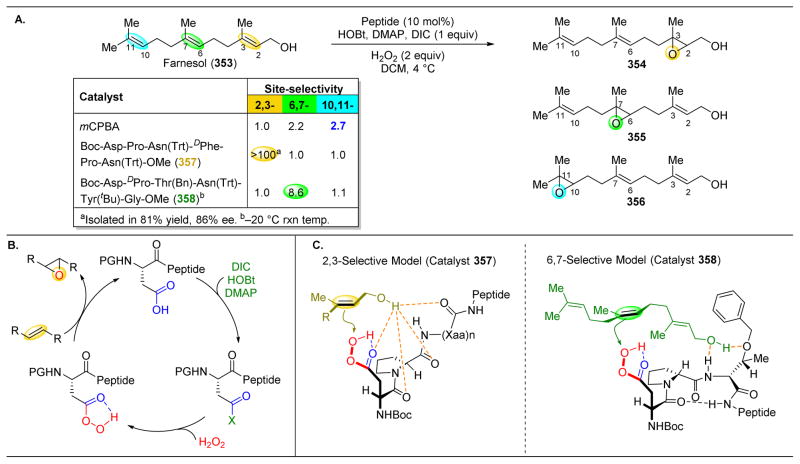
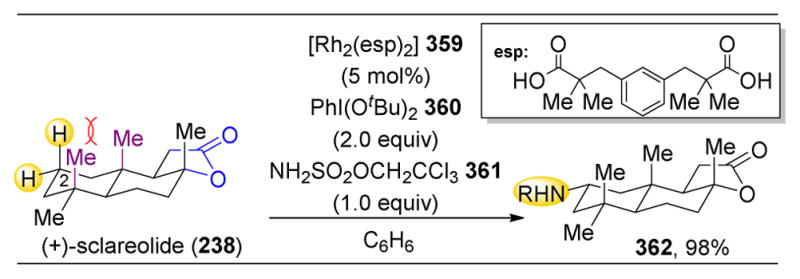
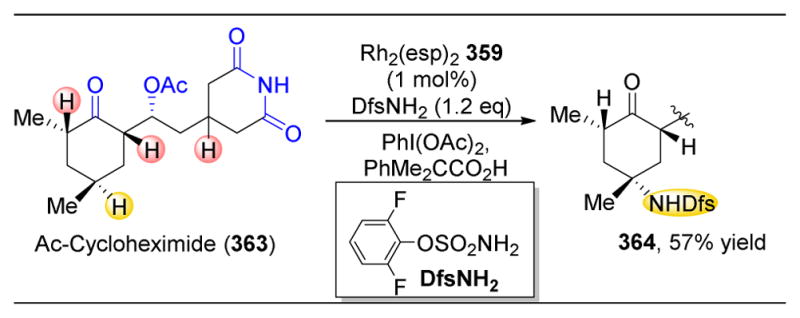
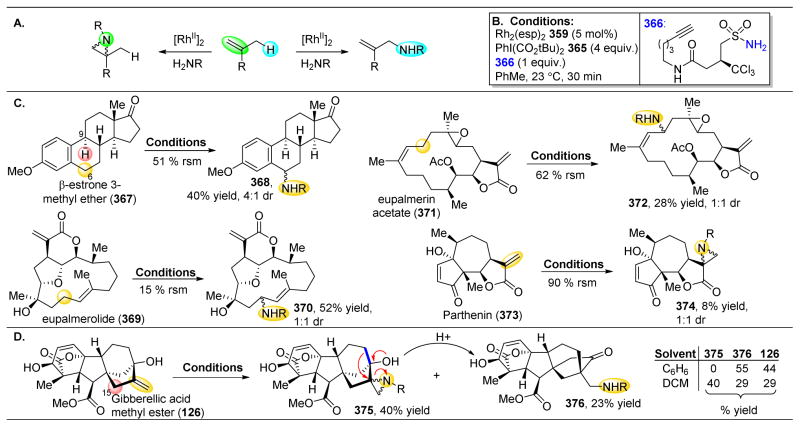
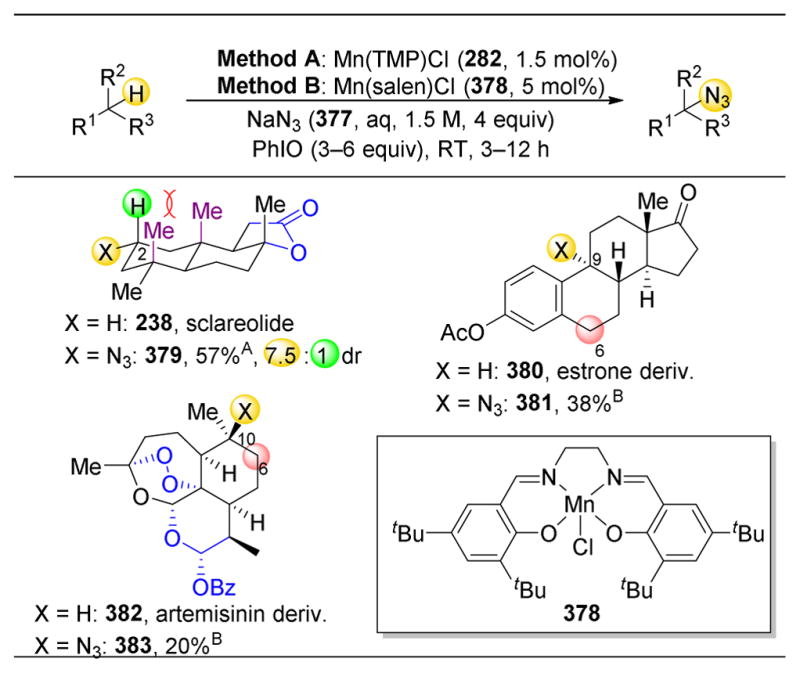
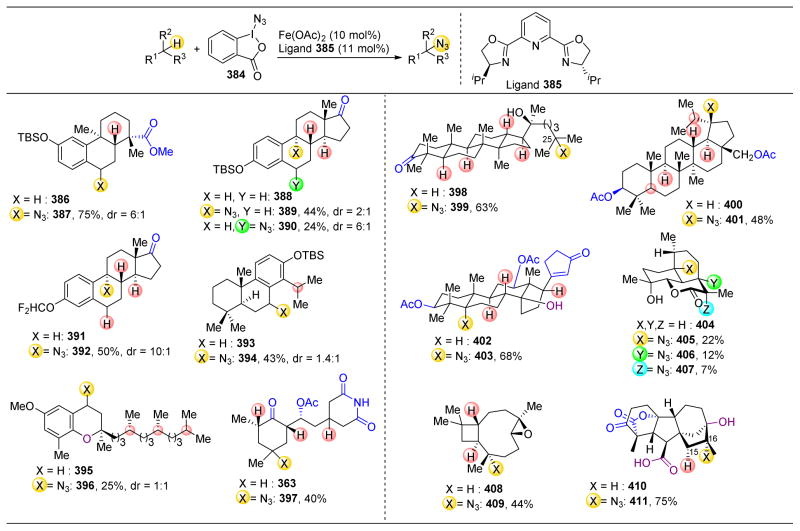
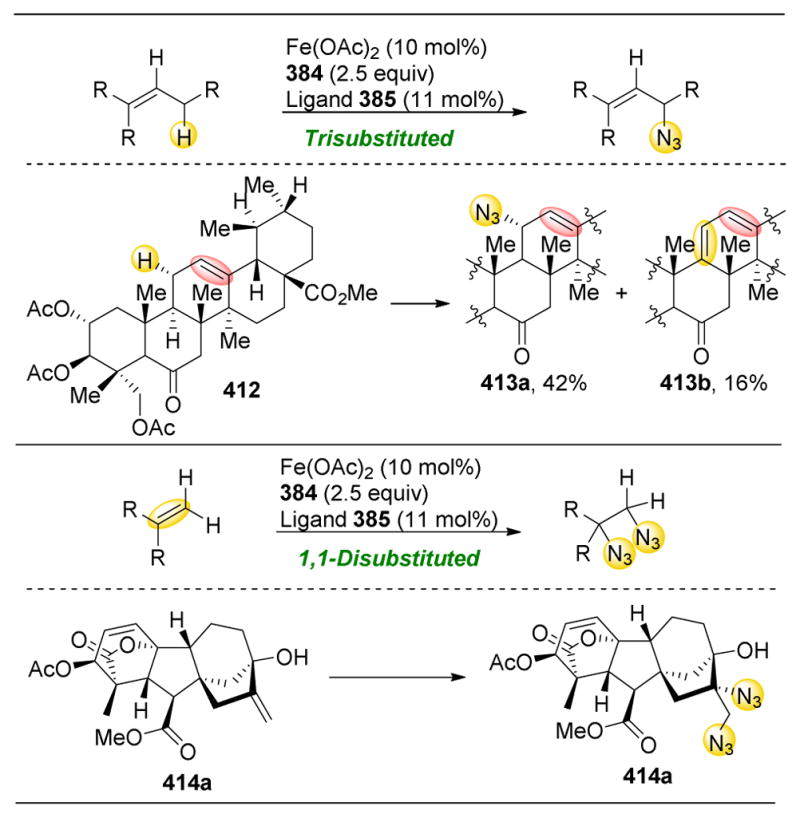
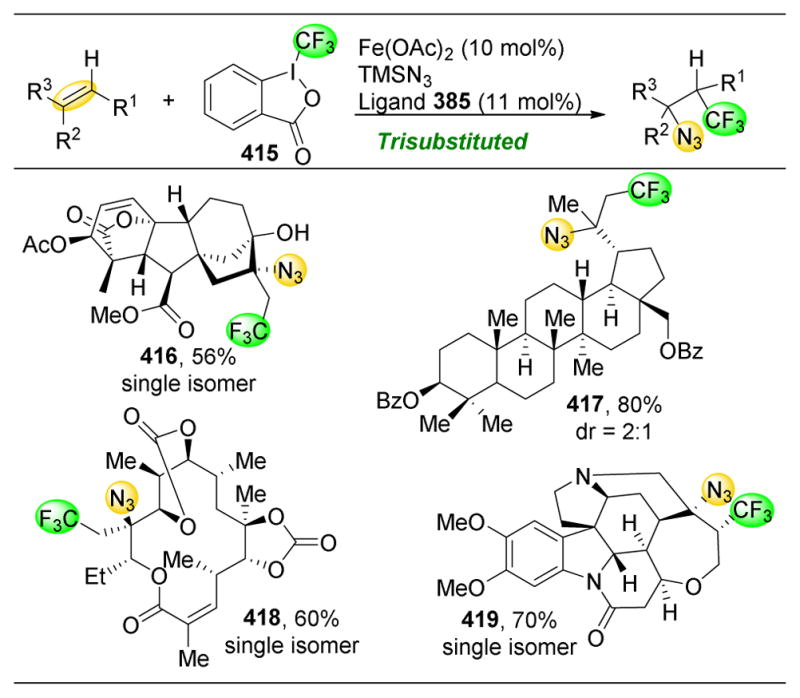
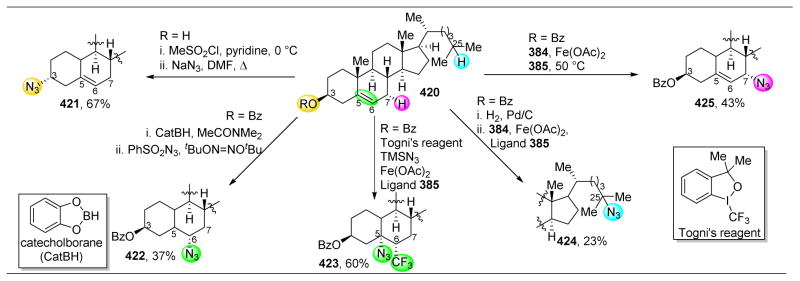
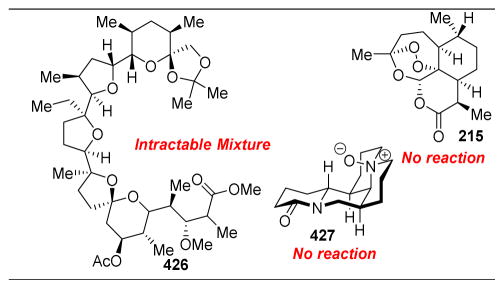
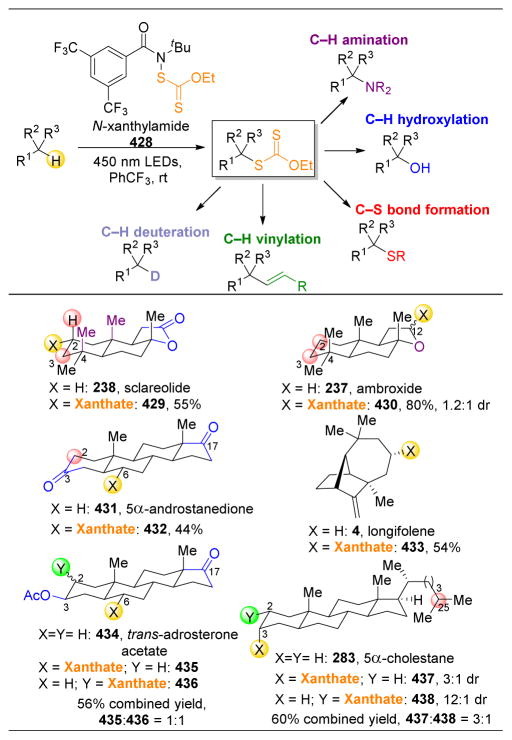
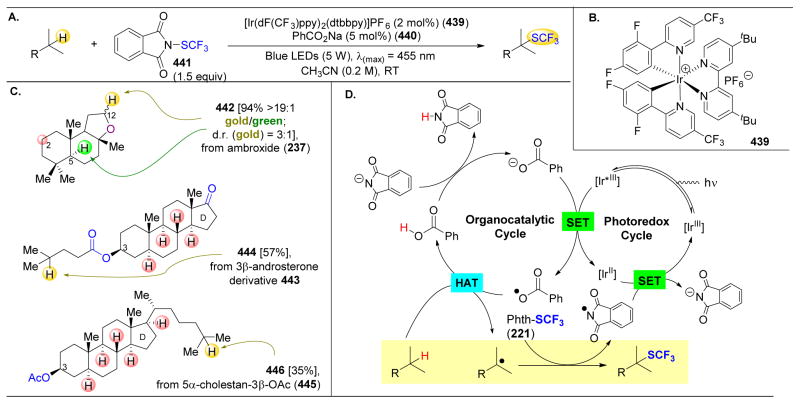
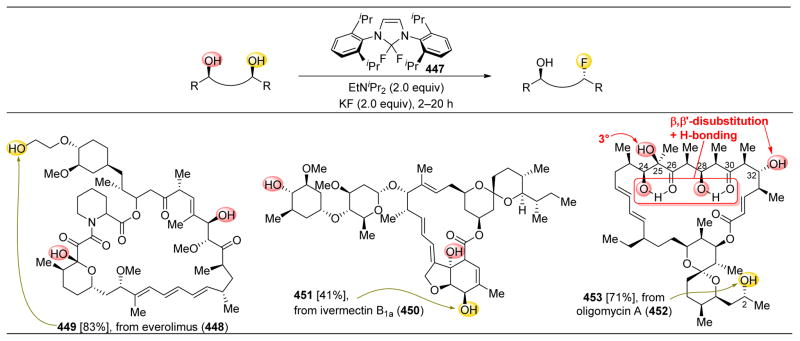
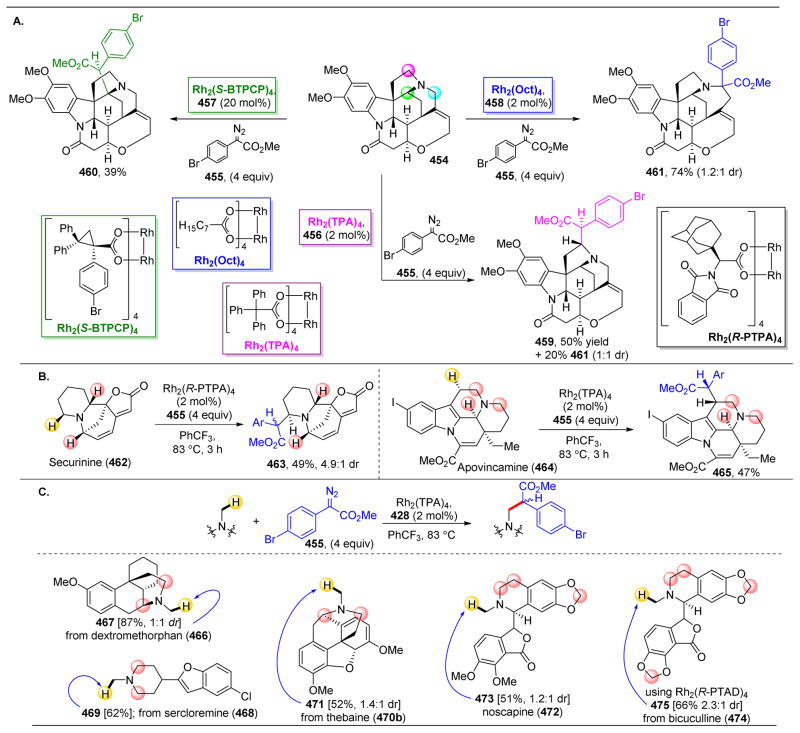
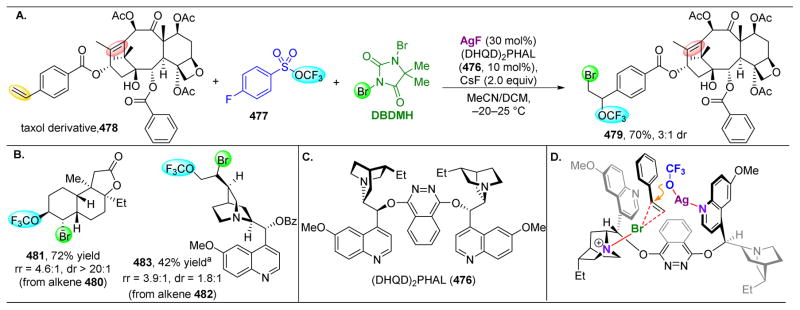
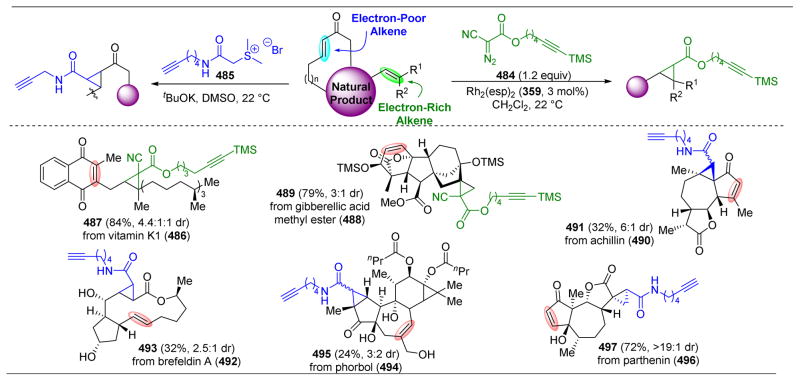
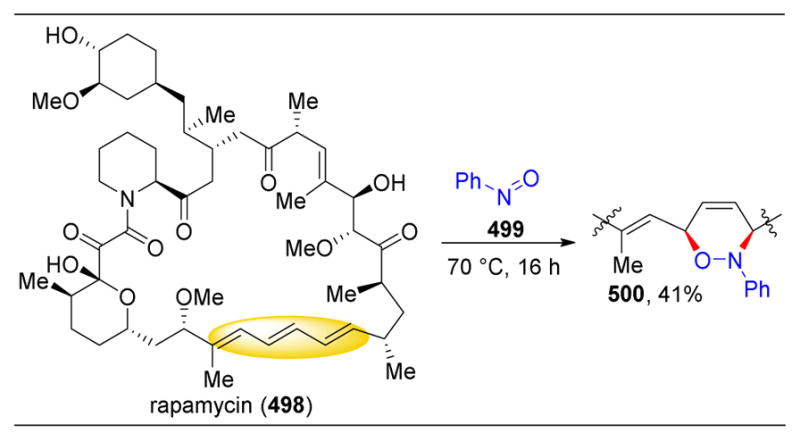
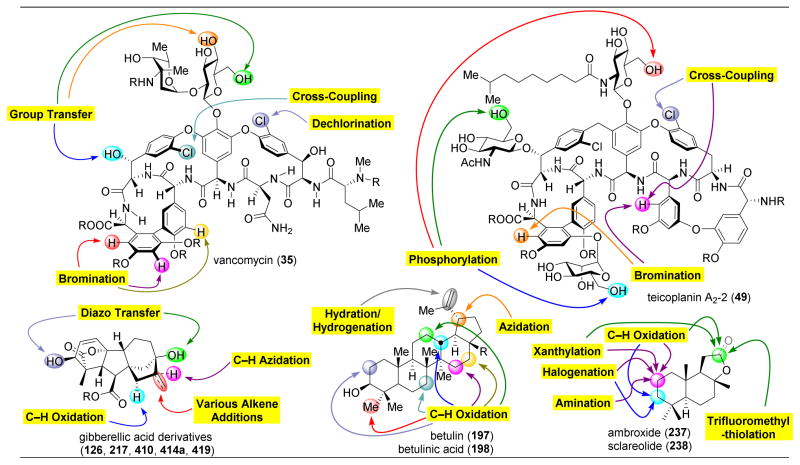
References
-
- Nicolaou KC, Montagnon T. Molecules That Changed the World. Wiley-VCH; 2008.
-
- Corey EJ, Kürti L, Czako B. Molecules and Medicine. Wiley-VCH; 2007.
-
- Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. - PubMed
-
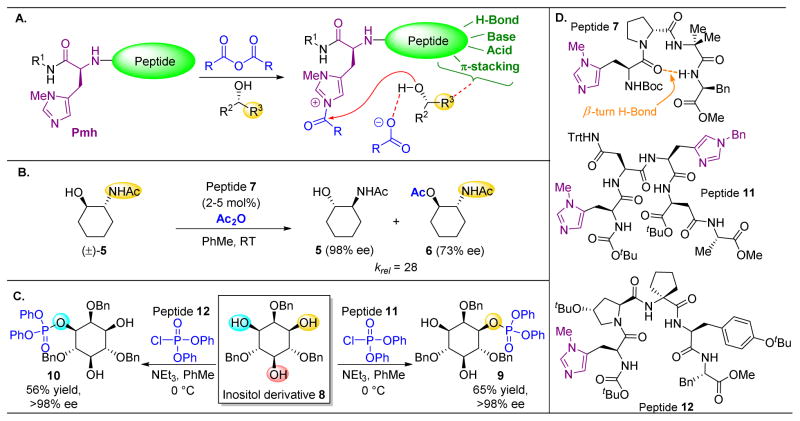
- Giuliano MW, Miller SJ. Site-Selective Reactions with Peptide-Based Catalysts. Top Curr Chem. 2016;372:157–202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
