

High-resolution cryo-EM proteasome structures in drug development
- PMID: 28580914
- PMCID: PMC5458494
- DOI: 10.1107/S2059798317007021
High-resolution cryo-EM proteasome structures in drug development
Abstract
With the recent advances in biological structural electron microscopy (EM), protein structures can now be obtained by cryo-EM and single-particle analysis at resolutions that used to be achievable only by crystallographic or NMR methods. We have explored their application to study protein-ligand interactions using the human 20S proteasome, a well established target for cancer therapy that is also being investigated as a target for an increasing range of other medical conditions. The map of a ligand-bound human 20S proteasome served as a proof of principle that cryo-EM is emerging as a realistic approach for more general structural studies of protein-ligand interactions, with the potential benefits of extending such studies to complexes that are unfavourable to other methods and allowing structure determination under conditions that are closer to physiological, preserving ligand specificity towards closely related binding sites. Subsequently, the cryo-EM structure of the Plasmodium falciparum 20S proteasome, with a new prototype specific inhibitor bound, revealed the molecular basis for the ligand specificity towards the parasite complex, which provides a framework to guide the development of highly needed new-generation antimalarials. Here, the cryo-EM analysis of the ligand-bound human and P. falciparum 20S proteasomes is reviewed, and a complete description of the methods used for structure determination is provided, including the strategy to overcome the bias orientation of the human 20S proteasome on electron-microscope grids and details of the icr3d software used for three-dimensional reconstruction.
Keywords: Plasmodium falciparum; cryo-EM; drug design; electron microscopy; human; icr3d; icr3dpro; inhibitors; malaria; proteasome; single particle.
Figures

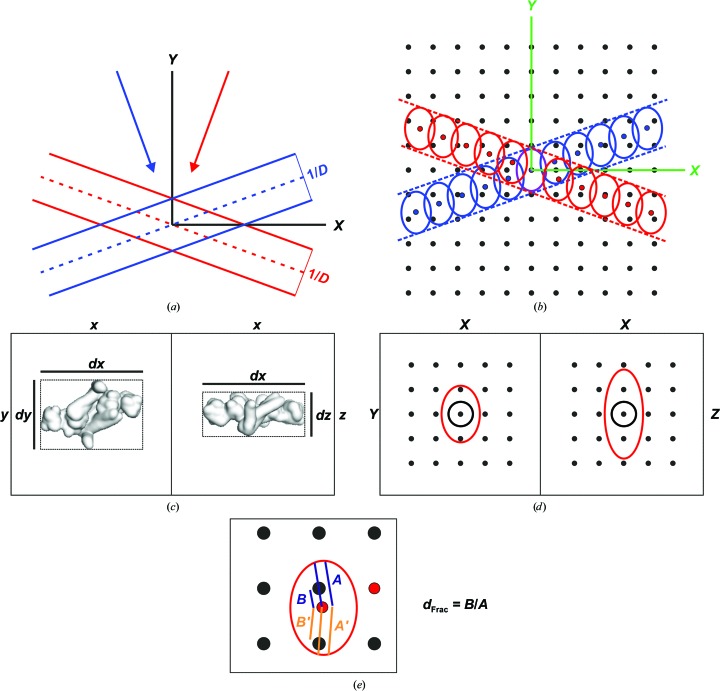
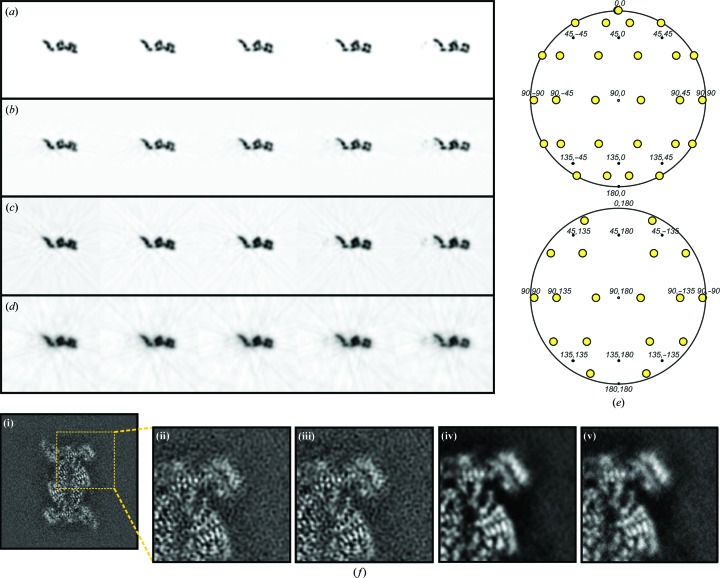
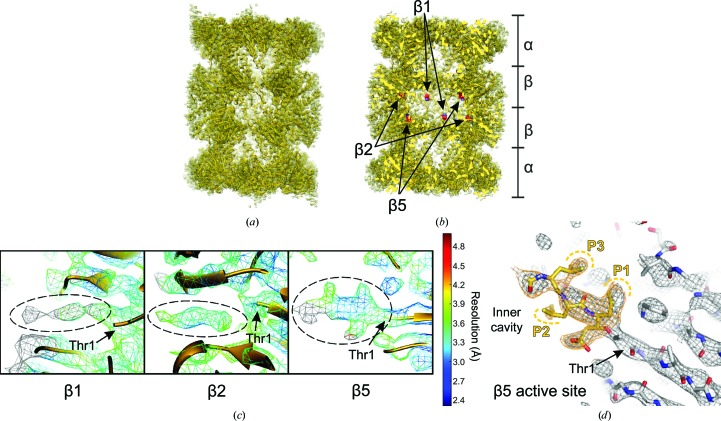
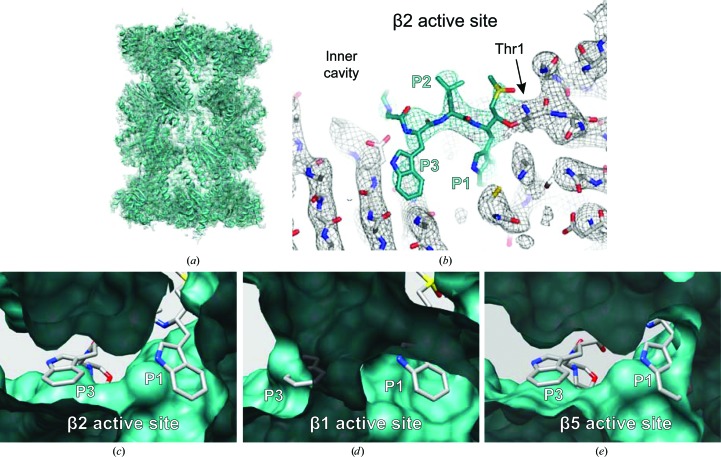
References
-
- Aebi, U. & Pollard, T. D. (1987). J. Electron Microsc. Tech. 7, 29–33. - PubMed
-
- Amos, L. A., Henderson, R. & Unwin, P. N. (1982). Prog. Biophys. Mol. Biol. 39, 183–231. - PubMed
-
- Ashley, E. A. et al. (2014). N. Engl. J. Med. 371, 411–423.
-
- Basler, M., Mundt, S., Bitzer, A., Schmidt, C. & Groettrup, M. (2015). Clin. Exp. Rheumatol. 33, S74–S79. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
